
Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells

Abstract
1. Introduction
2. Obesity, Adipose Tissue and Cardiovascular Diseases
3. Leptin—Friend or Foe in Atherosclerosis and Vascular Dysfunction
3.1. Clinical Studies
3.2. Animal Studies
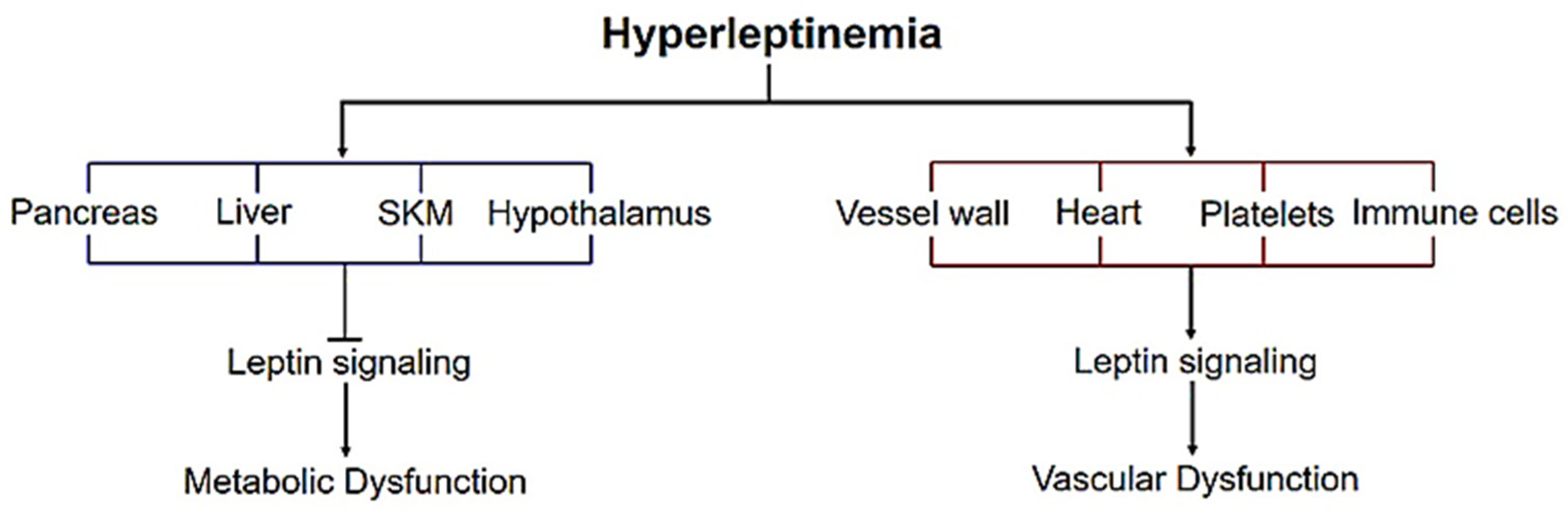
4. Leptin Signaling—Metabolic vs. Vascular Dysfunction
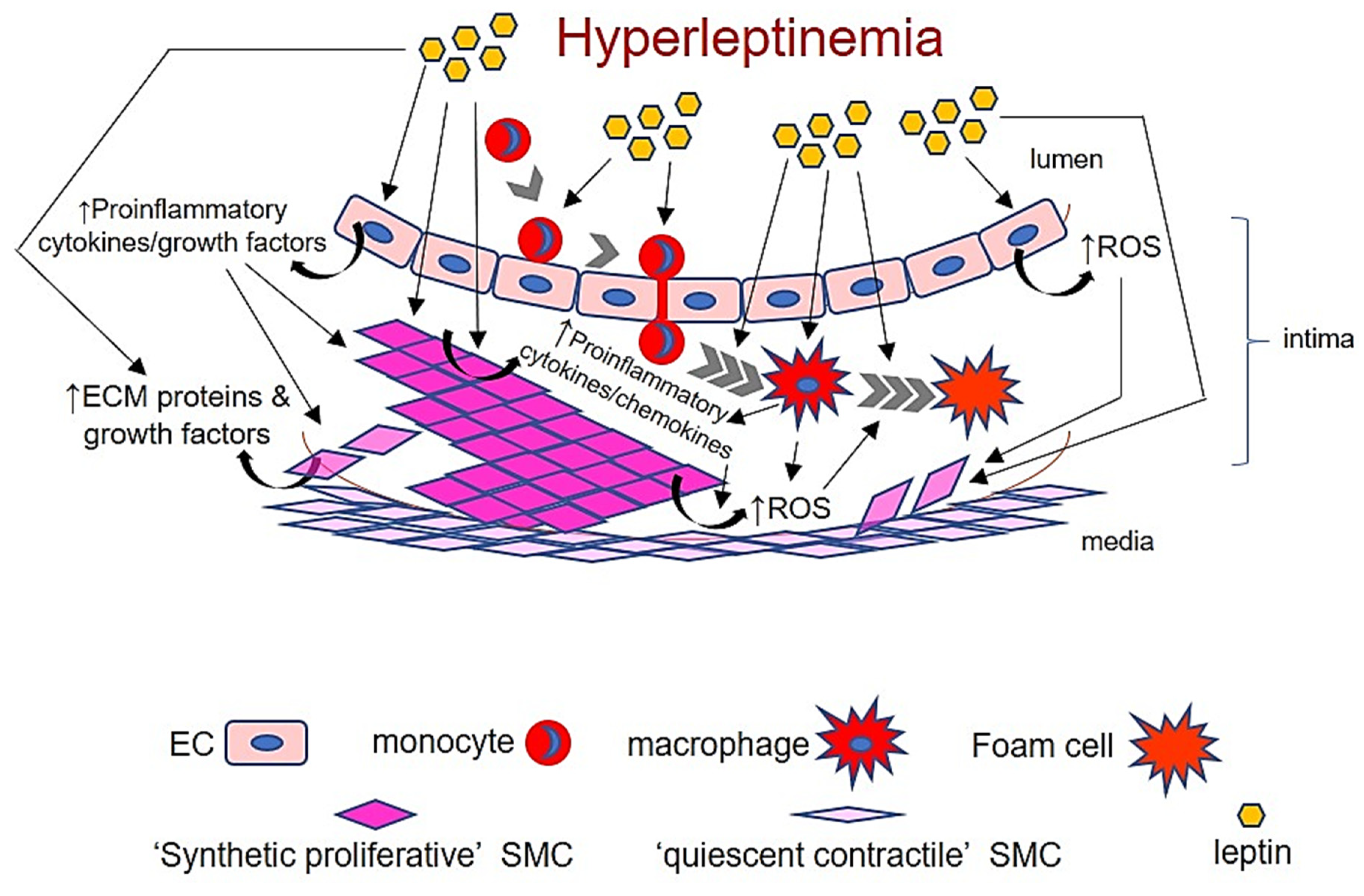
5. Basic Overview of Atherosclerosis
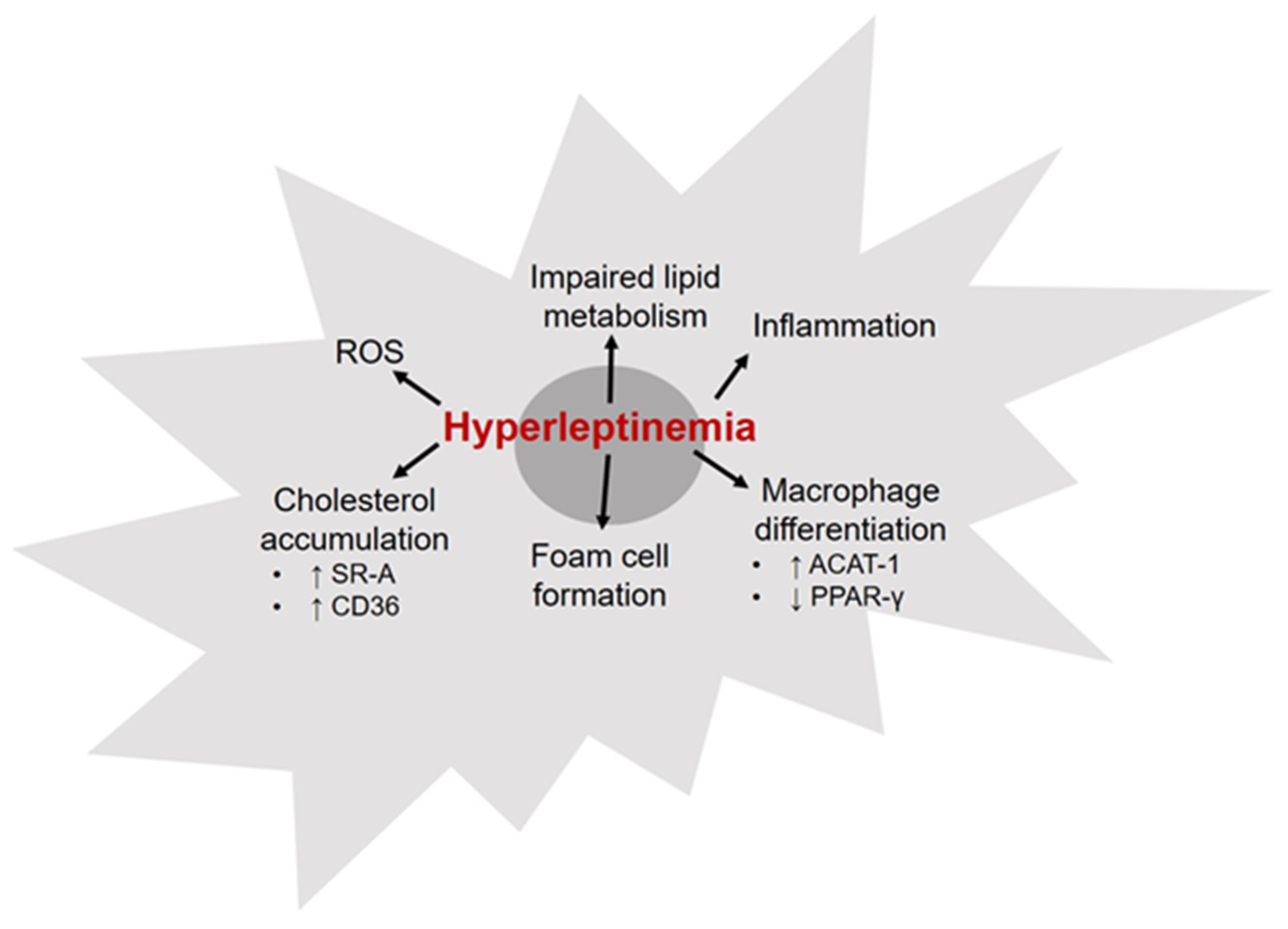
6. Leptin and Macrophages
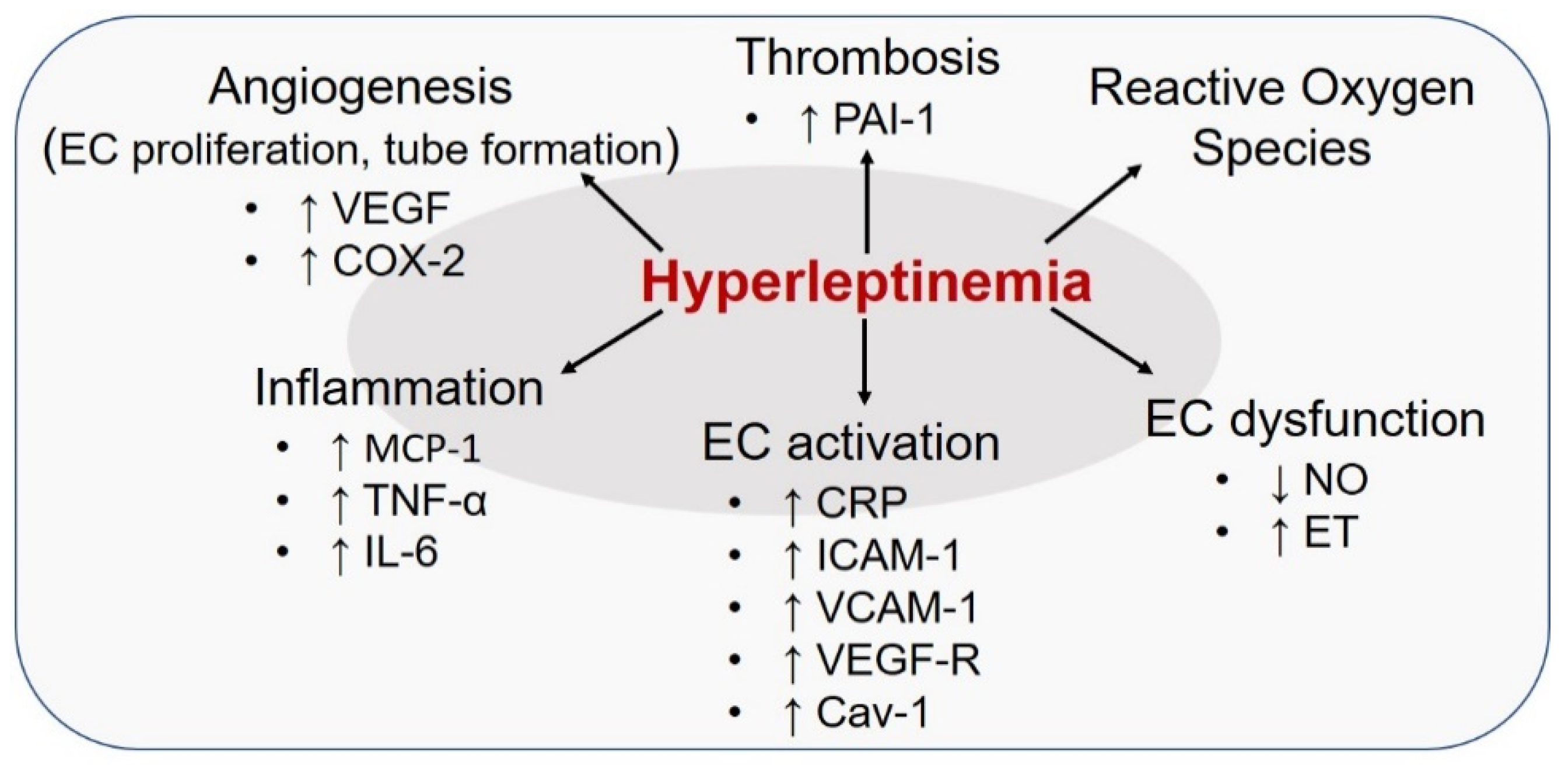
7. Leptin and Endothelial Cells
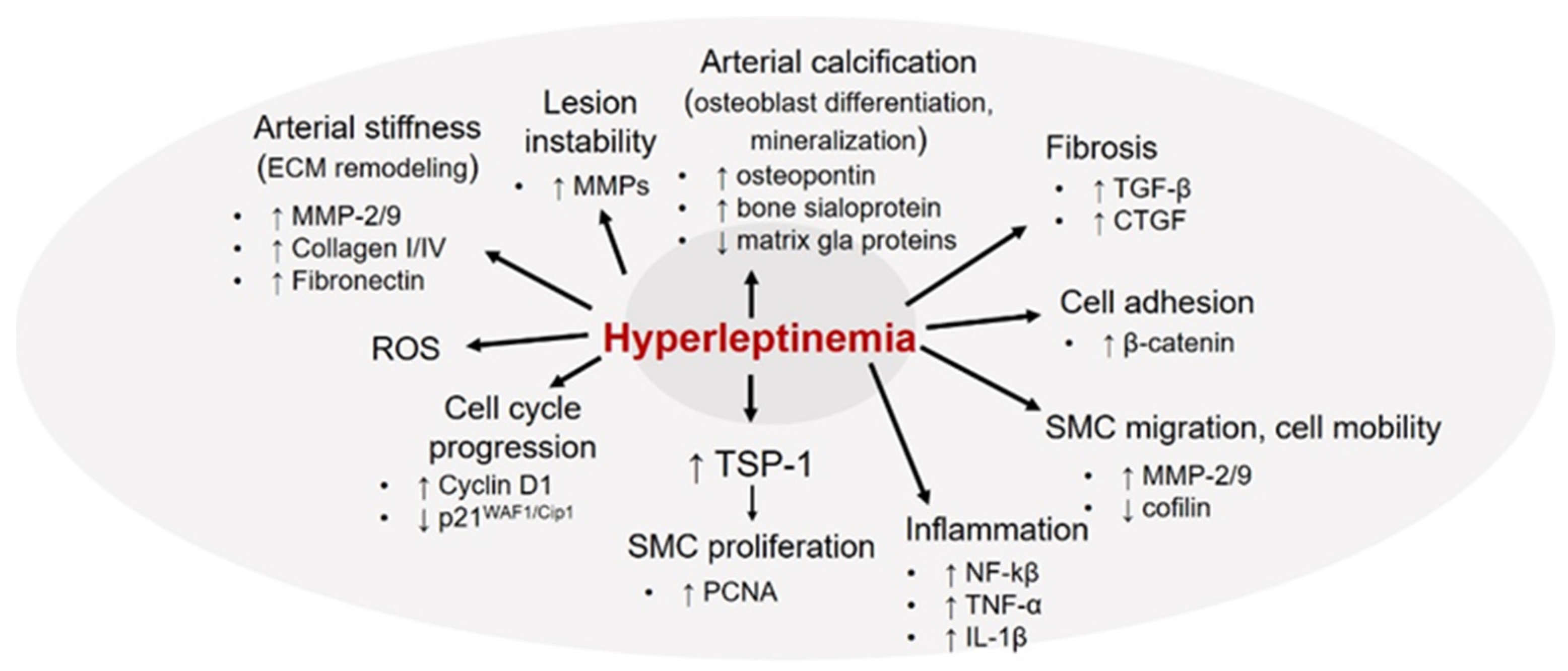
8. Leptin and Vascular Smooth Muscle Cells
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics-2021 update: A report from the american heart association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Juutilainen, A.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Similarity of the impact of type 1 and type 2 diabetes on cardiovascular mortality in middle-aged subjects. Diabetes Care 2008, 31, 714–719. [Google Scholar] [CrossRef]
- Barzilay, J.I.; Spiekerman, C.F.; Kuller, L.H.; Burke, G.L.; Bittner, V.; Gottdiener, J.S.; Brancati, F.L.; Orchard, T.J.; O’Leary, D.H.; Savage, P.J. Prevalence of clinical and isolated subclinical cardiovascular disease in older adults with glucose disorders: The Cardiovascular Health Study. Diabetes Care 2001, 24, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Creager, M.A.; Luscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef]
- Kanter, J.E.; Johansson, F.; LeBoeuf, R.C.; Bornfeldt, K.E. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ. Res. 2007, 100, 769–781. [Google Scholar] [CrossRef]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss. Arter. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef]
- Grundy, S.M. Obesity, metabolic syndrome, and coronary atherosclerosis. Circulation 2002, 105, 2696–2698. [Google Scholar] [CrossRef] [PubMed]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. 2014, 7, 587–591. [Google Scholar] [CrossRef]
- Abate, N. Obesity and cardiovascular disease. Pathogenetic role of the metabolic syndrome and therapeutic implications. J. Diabetes Complicat. 2000, 14, 154–174. [Google Scholar] [CrossRef]
- Isomaa, B.; Almgren, P.; Tuomi, T.; Forsen, B.; Lahti, K.; Nissen, M.; Taskinen, M.R.; Groop, L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care 2001, 24, 683–689. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Brit. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ragino, Y.I.; Stakhneva, E.M.; Polonskaya, Y.V.; Kashtanova, E.V. The role of secretory activity molecules of visceral adipocytes in abdominal obesity in the development of cardiovascular disease: A review. Biomolecules 2020, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef] [PubMed]
- Achike, F.I.; To, N.H.; Wang, H.; Kwan, C.Y. Obesity, metabolic syndrome, adipocytes and vascular function: A holistic viewpoint. Clin. Exp. Pharm. Physiol. 2011, 38, 1–10. [Google Scholar] [CrossRef]
- Konigorski, S.; Janke, J.; Drogan, D.; Bergmann, M.M.; Hierholzer, J.; Kaaks, R.; Boeing, H.; Pischon, T. Prediction of circulating adipokine levels based on body fat compartments and adipose tissue gene expression. Obes. Facts 2019, 12, 590–605. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Lau, D.C.; Dhillon, B.; Yan, H.; Szmitko, P.E.; Verma, S. Adipokines: Molecular links between obesity and atheroslcerosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2031–H2041. [Google Scholar] [CrossRef]
- Yoo, H.J.; Choi, K.M. Adipokines as a novel link between obesity and atherosclerosis. World J. Diabetes 2014, 5, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef] [PubMed]
- Denver, R.J.; Bonett, R.M.; Boorse, G.C. Evolution of leptin structure and function. Neuroendocrinology 2011, 94, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Wang, J.H.; Lee, C.J.; Hsu, B.G. Association between hyperleptinemia and cardiovascular outcomes in patients with coronary artery disease. Ther. Clin. Risk Manag. 2018, 14, 1855–1862. [Google Scholar] [CrossRef]
- Ren, J. Leptin and hyperleptinemia—From friend to foe for cardiovascular function. J. Endocrinol. 2004, 181, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Mazzone, T. Adipose tissue and atherosclerosis: Exploring the connection. Arter. Thromb. Vasc. Biol. 2007, 27, 996–1003. [Google Scholar] [CrossRef]
- Beltowski, J. Leptin and atherosclerosis. Atherosclerosis 2006, 189, 47–60. [Google Scholar] [CrossRef]
- Reilly, M.P.; Iqbal, N.; Schutta, M.; Wolfe, M.L.; Scally, M.; Localio, A.R.; Rader, D.J.; Kimmel, S.E. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3872–3878. [Google Scholar] [CrossRef]
- McMahon, M.; Skaggs, B.J.; Sahakian, L.; Grossman, J.; FitzGerald, J.; Ragavendra, N.; Charles-Schoeman, C.; Chernishof, M.; Gorn, A.; Witztum, J.L.; et al. High plasma leptin levels confer increased risk of atherosclerosis in women with systemic lupus erythematosus, and are associated with inflammatory oxidised lipids. Ann. Rheum. Dis. 2011, 70, 1619–1624. [Google Scholar] [CrossRef]
- Montazerifar, F.; Bolouri, A.; Paghalea, R.S.; Mahani, M.K.; Karajibani, M. Obesity, serum resistin and leptin levels linked to coronary artery disease. Arq. Bras. Cardiol. 2016, 107, 348–353. [Google Scholar] [CrossRef]
- Rahmani, A.; Toloueitabar, Y.; Mohsenzadeh, Y.; Hemmati, R.; Sayehmiri, K.; Asadollahi, K. Association between plasma leptin/adiponectin ratios with the extent and severity of coronary artery disease. BMC Cardiovasc. Dis. 2020, 20, 474. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Shen, Y.; Lin, B.; Li, Y.; He, X.; Zhang, Y.; Lu, L.; Shen, W.; Zhang, Q.; et al. Association between serum leptin level and calcific aortic valve disease. J. Am. Heart Assoc. 2019, 8, e012495. [Google Scholar] [CrossRef]
- Khafaji, H.A.; Bener, A.B.; Rizk, N.M.; Al Suwaidi, J. Elevated serum leptin levels in patients with acute myocardial infarction; correlation with coronary angiographic and echocardiographic findings. BMC Res. Notes 2012, 5, 262. [Google Scholar] [CrossRef] [PubMed]
- Wallerstedt, S.M.; Eriksson, A.L.; Niklason, A.; Ohlsson, C.; Hedner, T. Serum leptin and myocardial infarction in hypertension. Blood Press. 2004, 13, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guo, W.; Li, J.; Cao, S.; Zhang, J.; Pan, J.; Wang, Z.; Wen, P.; Shi, X.; Zhang, S. Leptin concentration and risk of coronary heart disease and stroke: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0166360. [Google Scholar] [CrossRef] [PubMed]
- Ahiante, B.O.; Smith, W.; Lammertyn, L.; Schutte, A.E. Leptin and the vasculature in young adults: The African-PREDICT study. Eur. J. Clin. Investig. 2019, 49, e13039. [Google Scholar] [CrossRef]
- Schafer, K.; Halle, M.; Goeschen, C.; Dellas, C.; Pynn, M.; Loskutoff, D.J.; Konstantinides, S. Leptin promotes vascular remodeling and neointimal growth in mice. Arter. Thromb. Vasc. Biol. 2004, 24, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Nguyen, T.B.; Totary-Jain, H.; Dansky, H.; Marx, S.O.; Marks, A.R. Leptin-enhanced neointimal hyperplasia is reduced by mTOR and PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 19006–19011. [Google Scholar] [CrossRef]
- Schroeter, M.R.; Eschholz, N.; Herzberg, S.; Jerchel, I.; Leifheit-Nestler, M.; Czepluch, F.S.; Chalikias, G.; Konstantinides, S.; Schafer, K. Leptin-dependent and leptin-independent paracrine effects of perivascular adipose tissue on neointima formation. Arter. Thromb. Vasc. Biol. 2013, 33, 980–987. [Google Scholar] [CrossRef]
- Stephenson, K.; Tunstead, J.; Tsai, A.; Gordon, R.; Henderson, S.; Dansky, H.M. Neointimal formation after endovascular arterial injury is markedly attenuated in db/db mice. Arter. Thromb. Vasc. Biol. 2003, 23, 2027–2033. [Google Scholar] [CrossRef][Green Version]
- Bodary, P.F.; Shen, Y.; Ohman, M.; Bahrou, K.L.; Vargas, F.B.; Cudney, S.S.; Wickenheiser, K.J.; Myers, M.G., Jr.; Eitzman, D.T. Leptin regulates neointima formation after arterial injury through mechanisms independent of blood pressure and the leptin receptor/STAT3 signaling pathways involved in energy balance. Arter. Thromb. Vasc. Biol. 2007, 27, 70–76. [Google Scholar] [CrossRef]
- Schroeter, M.R.; Leifheit-Nestler, M.; Hubert, A.; Schumann, B.; Gluckermann, R.; Eschholz, N.; Kruger, N.; Lutz, S.; Hasenfuss, G.; Konstantinides, S.; et al. Leptin promotes neointima formation and smooth muscle cell proliferation via NADPH oxidase activation and signalling in caveolin-rich microdomains. Cardiovasc. Res. 2013, 99, 555–565. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bodary, P.F.; Gu, S.; Shen, Y.; Hasty, A.H.; Buckler, J.M.; Eitzman, D.T. Recombinant leptin promotes atherosclerosis and thrombosis in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2005, 25, e119–e122. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Shinozaki, S.; Nakazawa, T.; Kawakami, A.; Ai, M.; Kaneko, E.; Kitagawa, M.; Kondo, K.; Chait, A.; Shimokado, K. Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis 2008, 196, 68–75. [Google Scholar] [CrossRef]
- Taleb, S.; Herbin, O.; Ait-Oufella, H.; Verreth, W.; Gourdy, P.; Barateau, V.; Merval, R.; Esposito, B.; Clement, K.; Holvoet, P.; et al. Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 2691–2698. [Google Scholar] [CrossRef]
- Ganguly, R.; Khanal, S.; Mathias, A.; Gupta, S.; Lallo, J.; Sahu, S.; Ohanyan, V.; Patel, A.; Storm, K.; Datta, S.; et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE(-/-) Mice Against Leptin-Induced Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e112–e127. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Wu, T.J.; Chin, J.; Mitnaul, L.J.; Hernandez, M.; Cai, T.Q.; Ren, N.; Waters, M.G.; Wright, S.D.; Cheng, K. Increased hypercholesterolemia and atherosclerosis in mice lacking both ApoE and leptin receptor. Atherosclerosis 2005, 181, 251–259. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ebert, T.; Kloting, N.; Dokas, J.; Jeromin, F.; Jessnitzer, B.; Burkhardt, R.; Fasshauer, M.; Kralisch, S. Leptin dose-dependently decreases atherosclerosis by attenuation of hypercholesterolemia and induction of adiponectin. Biochim. Biophys. Acta 2016, 1862, 113–120. [Google Scholar] [CrossRef]
- Jun, J.Y.; Ma, Z.; Pyla, R.; Segar, L. Leptin treatment inhibits the progression of atherosclerosis by attenuating hypercholesterolemia in type 1 diabetic Ins2(+/Akita): ApoE(−/−) mice. Atherosclerosis 2012, 225, 341–347. [Google Scholar] [CrossRef][Green Version]
- Park, H.K.; Ahima, R.S. Leptin signaling. F1000prime Rep. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Tintut, Y.; Ballard, A.; Fogelman, A.M.; Demer, L.L. Leptin enhances the calcification of vascular cells: Artery wall as a target of leptin. Circ. Res. 2001, 88, 954–960. [Google Scholar] [CrossRef]
- Kang, S.M.; Kwon, H.M.; Hong, B.K.; Kim, D.; Kim, I.J.; Choi, E.Y.; Jang, Y.; Kim, H.S.; Kim, M.S.; Kwon, H.C. Expression of leptin receptor (Ob-R) in human atherosclerotic lesions: Potential role in intimal neovascularization. YONSEI Med. J. 2000, 41, 68–75. [Google Scholar] [CrossRef]
- Schroeter, M.R.; Schneiderman, J.; Schumann, B.; Gluckermann, R.; Grimmas, P.; Buchwald, A.B.; Tirilomis, T.; Schondube, F.A.; Konstantinides, S.V.; Schafer, K. Expression of the leptin receptor in different types of vascular lesions. Histochem. Cell Biol. 2007, 128, 323–333. [Google Scholar] [CrossRef]
- Gonzalez, R.R.; Cherfils, S.; Escobar, M.; Yoo, J.H.; Carino, C.; Styer, A.K.; Sullivan, B.T.; Sakamoto, H.; Olawaiye, A.; Serikawa, T.; et al. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2). J. Biol. Chem. 2006, 281, 26320–26328. [Google Scholar] [CrossRef]
- Cha, J.J.; Hyun, Y.Y.; Jee, Y.H.; Lee, M.J.; Han, K.H.; Kang, Y.S.; Han, S.Y.; Cha, D.R. Plasma leptin concentrations are greater in type II diabetic patients and stimulate monocyte chemotactic peptide-1 synthesis via the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway. Kidney Res. Clin. Pract. 2012, 31, 177–185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leung, J.C.; Chan, L.Y.; Tang, S.C.; Chu, K.M.; Lai, K.N. Leptin induces TGF-beta synthesis through functional leptin receptor expressed by human peritoneal mesothelial cell. Kidney Int. 2006, 69, 2078–2086. [Google Scholar] [CrossRef]
- Li, L.; Mamputu, J.C.; Wiernsperger, N.; Renier, G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: Inhibitory effect of metformin. Diabetes 2005, 54, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kwon, H.M.; Lim, H.J.; Hong, B.K.; Lee, J.Y.; Park, B.E.; Jang, Y.; Cho, S.Y.; Kim, H.S. Potential role of leptin in angiogenesis: Leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp. Mol. Med. 2001, 33, 95–102. [Google Scholar] [CrossRef]
- Quehenberger, P.; Exner, M.; Sunder-Plassmann, R.; Ruzicka, K.; Bieglmayer, C.; Endler, G.; Muellner, C.; Speiser, W.; Wagner, O. Leptin induces endothelin-1 in endothelial cells in vitro. Circ. Res. 2002, 90, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Kaneda, Y.; Guzman, M.; Brownlee, M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J. Biol. Chem. 2001, 276, 25096–25100. [Google Scholar] [CrossRef]
- Chavez, R.J.; Haney, R.M.; Cuadra, R.H.; Ganguly, R.; Adapala, R.K.; Thodeti, C.K.; Raman, P. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am. J. Physiol. Cell Physiol. 2012, 303, C179–C191. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Rovella, V.; Di Daniele, N.; Mores, N.; Veneziani, A.; Cardillo, C. Leptin stimulates both endothelin-1 and nitric oxide activity in lean subjects but not in patients with obesity-related metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Knudson, J.D.; Dincer, U.D.; Zhang, C.; Swafford, A.N.; Jr Koshida, R.; Picchi, A.; Focardi, M.; Dick, G.M.; Tune, J.D. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H48–H56. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, D.; Paletas, K.; Koliakos, G.; Kaloyianni, M. The ambiguous role of the Na+-H+ exchanger isoform 1 (NHE1) in leptin-induced oxidative stress in human monocytes. Cell Stress Chaperones 2009, 14, 591–601. [Google Scholar] [CrossRef]
- Buis, D.T.P.; Christen, T.; Smit, R.A.J.; de Mutsert, R.; Jukema, J.W.; Cannegieter, S.C.; Lijfering, W.M.; Rosendaal, F.R. The association between leptin concentration and blood coagulation: Results from the NEO study. Thromb. Res. 2020, 188, 44–48. [Google Scholar] [CrossRef]
- Petrini, S.; Neri, T.; Lombardi, S.; Cordazzo, C.; Balia, C.; Scalise, V.; Paggiaro, P.; Pedrinelli, R.; Celi, A. Leptin induces the generation of procoagulant, tissue factor bearing microparticles by human peripheral blood mononuclear cells. Biochim. Biophys. Acta 2016, 1860, 1354–1361. [Google Scholar] [CrossRef]
- Pieterse, C.; Schutte, R.; Schutte, A.E. Leptin links with plasminogen activator inhibitor-1 in human obesity: The SABPA study. Hypertens. Res. 2015, 38, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Ben-Aicha, S.; Badimon, L. New insights into the role of adipose tissue in thrombosis. Cardiovasc. Res. 2017, 113, 1046–1054. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Paivarinta, U.; Moilanen, T.; Moilanen, E. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage—Mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediat. Inflamm. 2009, 2009, 345838. [Google Scholar] [CrossRef]
- Tahergorabi, Z.; Khazaei, M. Leptin and its cardiovascular effects: Focus on angiogenesis. Adv. Biomed. Res. 2015, 4, 79. [Google Scholar] [PubMed]
- Sanches, P.L.; de Mello, M.T.; Elias, N.; Fonseca, F.A.; Campos, R.M.; Carnier, J.; de Piano, A.; Masquio, D.C.; Silva, P.L.; Oyama, L.M.; et al. Hyperleptinemia: Implications on the inflammatory state and vascular protection in obese adolescents submitted to an interdisciplinary therapy. Inflammation 2014, 37, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovas. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Choudhury, R.P.; Lee, J.M.; Greaves, D.R. Mechanisms of disease: Macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 309–315. [Google Scholar] [CrossRef]
- Webb, N.R.; Moore, K.J. Macrophage-derived foam cells in atherosclerosis: Lessons from murine models and implications for therapy. Curr. Drug Targets 2007, 8, 1249–1263. [Google Scholar] [CrossRef]
- Fadini, G.P.; Simoni, F.; Cappellari, R.; Vitturi, N.; Galasso, S.; Vigili de Kreutzenberg, S.; Previato, L.; Avogaro, A. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis 2014, 237, 805–808. [Google Scholar] [CrossRef]
- Lee, M.K.; Moore, X.L.; Fu, Y.; Al-Sharea, A.; Dragoljevic, D.; Fernandez-Rojo, M.A.; Parton, R.; Sviridov, D.; Murphy, A.J.; Chin-Dusting, J.P. High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br. J. Pharm. 2016, 173, 741–751. [Google Scholar] [CrossRef]
- Gruen, M.L.; Hao, M.; Piston, D.W.; Hasty, A.H. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am. J. Physiol. Cell Physiol. 2007, 293, C1481–C1488. [Google Scholar] [CrossRef] [PubMed]
- Kjerrulf, M.; Berke, Z.; Aspegren, A.; Umaerus, M.; Nilsson, T.; Svensson, L.; Hurt-Camejo, E. Reduced cholesterol accumulation by leptin deficient (ob/ob) mouse macrophages. Inflamm. Res. 2006, 55, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Tsiotra, P.C.; Boutati, E.; Dimitriadis, G.; Raptis, S.A. High insulin and leptin increase resistin and inflammatory cytokine production from human mononuclear cells. Biomed Res. Int. 2013, 2013, 487081. [Google Scholar] [CrossRef]
- Hongo, S.; Watanabe, T.; Arita, S.; Kanome, T.; Kageyama, H.; Shioda, S.; Miyazaki, A. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E474–E482. [Google Scholar] [CrossRef]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Cabrero, A.; Cubero, M.; Llaverias, G.; Alegret, M.; Sanchez, R.; Laguna, J.C.; Vazquez-Carrera, M. Leptin down-regulates peroxisome proliferator-activated receptor gamma (PPAR-gamma) mRNA levels in primary human monocyte-derived macrophages. Mol. Cell. Biochem. 2005, 275, 173–179. [Google Scholar] [CrossRef]
- Surmi, B.K.; Atkinson, R.D.; Gruen, M.L.; Coenen, K.R.; Hasty, A.H. The role of macrophage leptin receptor in aortic root lesion formation. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E488–E495. [Google Scholar] [CrossRef]
- Payne, G.A.; Tune, J.D.; Knudson, J.D. Leptin-induced endothelial dysfunction: A target for therapeutic interventions. Curr. Pharm. Des. 2014, 20, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Knudson, J.D.; Payne, G.A.; Borbouse, L.; Tune, J.D. Leptin and mechanisms of endothelial dysfunction and cardiovascular disease. Curr. Hypertens. Rep. 2008, 10, 434–439. [Google Scholar] [CrossRef]
- Kwaifa, I.K.; Bahari, H.; Yong, Y.K.; Noor, S.M. Endothelial dysfunction in obesity-induced inflammation: Molecular mechanisms and clinical implications. Biomolecules 2020, 10, 291. [Google Scholar] [CrossRef]
- Beltowski, J. Leptin and the regulation of endothelial function in physiological and pathological conditions. Clin. Exp. Pharm. Physiol. 2012, 39, 168–178. [Google Scholar] [CrossRef]
- Manuel-Apolinar, L.; Lopez-Romero, R.; Zarate, A.; Damasio, L.; Ruiz, M.; Castillo-Hernandez, C.; Guevara, G.; Mera-Jimenez, E. Leptin mediated ObRb receptor increases expression of adhesion intercellular molecules and cyclooxygenase 2 on murine aorta tissue inducing endothelial dysfunction. Int. J. Clin. Exper. Med. 2013, 6, 192–196. [Google Scholar]
- Singh, P.; Hoffmann, M.; Wolk, R.; Shamsuzzaman, A.S.; Somers, V.K. Leptin induces C-reactive protein expression in vascular endothelial cells. Arter. Thromb. Vasc. Biol. 2007, 27, e302–e307. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef]
- Jamroz-Wisniewska, A.; Gertler, A.; Solomon, G.; Wood, M.E.; Whiteman, M.; Beltowski, J. Leptin-induced endothelium-dependent vasorelaxation of peripheral arteries in lean and obese rats: Role of nitric oxide and hydrogen sulfide. PLoS ONE 2014, 9, e86744. [Google Scholar] [CrossRef] [PubMed]
- Benkhoff, S.; Loot, A.E.; Pierson, I.; Sturza, A.; Kohlstedt, K.; Fleming, I.; Shimokawa, H.; Grisk, O.; Brandes, R.P.; Schroder, K. Leptin potentiates endothelium-dependent relaxation by inducing endothelial expression of neuronal NO synthase. Arter. Thromb. Vasc. Biol. 2012, 32, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Peterson, T.E.; Sert-Kuniyoshi, F.H.; Jensen, M.D.; Somers, V.K. Leptin upregulates caveolin-1 expression: Implications for development of atherosclerosis. Atherosclerosis 2011, 217, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Garonna, E.; Botham, K.M.; Birdsey, G.M.; Randi, A.M.; Gonzalez-Perez, R.R.; Wheeler-Jones, C.P. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS ONE 2011, 6, e18823. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Peterson, T.E.; Barber, K.R.; Kuniyoshi, F.S.; Jensen, A.; Hoffmann, M.; Shamsuzzaman, A.S.; Somers, V.K. Leptin upregulates the expression of plasminogen activator inhibitor-1 in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 392, 47–52. [Google Scholar] [CrossRef]
- Cirillo, P.; Angri, V.; De Rosa, S.; Cali, G.; Petrillo, G.; Maresca, F.; D’Ascoli, G.L.; Maietta, P.; Brevetti, L.; Chiariello, M. Pro-atherothrombotic effects of leptin in human coronary endothelial cells. Thromb. Haemost. 2010, 103, 1065–1075. [Google Scholar]
- Hubert, A.; Bochenek, M.L.; Schutz, E.; Gogiraju, R.; Munzel, T.; Schafer, K. Selective deletion of leptin signaling in endothelial cells enhances neointima formation and phenocopies the vascular effects of diet-induced obesity in mice. Arter. Thromb. Vasc. Biol. 2017, 37, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Xiong, X.; Wang, H.; You, S.; Zeng, H. Leptin-induced vascular smooth muscle cell proliferation via regulating cell cycle, activating ERK1/2 and NF-kappaB. Acta Biochim. Biophys. Sin. 2010, 42, 325–331. [Google Scholar] [CrossRef]
- Noblet, J.N.; Goodwill, A.G.; Sassoon, D.J.; Kiel, A.M.; Tune, J.D. Leptin augments coronary vasoconstriction and smooth muscle proliferation via a Rho-kinase-dependent pathway. Basic. Res. Cardiol. 2016, 111, 25. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Leu, S.Y.; Peng, Y.J.; Lee, Y.M.; Hsu, C.H.; Chou, S.C.; Yen, M.H.; Cheng, P.Y. Genistein suppresses leptin-induced proliferation and migration of vascular smooth muscle cells and neointima formation. J. Cell. Mol. Med. 2017, 21, 422–431. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Lee, Y.M.; Hsu, C.H.; Leu, S.Y.; Chiang, H.Y.; Yen, M.H.; Cheng, P.Y. The effect of ferulic acid ethyl ester on leptin-induced proliferation and migration of aortic smooth muscle cells. Exp. Mol. Med. 2015, 47, e180. [Google Scholar] [CrossRef]
- Hariri, M.A.; Jaffa, M.A.; Saoud, R.; Zhao, J.; Zhu, R.; Jaffa, A.A.; El-Achkar, G.A.; Moussa, M.; Kobeissy, F.; Hassan, A.; et al. Vascular cells proteome associated with bradykinin and leptin inflammation and oxidative stress signals. Antioxidants 2020, 9, 1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, M.; Yin, L.; Fu, G.; Liu, Z. Role of thrombospondin1 and thrombospondin2 in cardiovascular diseases (Review). Int. J. Mol. Med. 2020, 45, 1275–1293. [Google Scholar]
- Stenina, O.I.; Plow, E.F. Counterbalancing forces: What is thrombospondin-1 doing in atherosclerotic lesions? Circ. Res. 2008, 103, 1053–1055. [Google Scholar] [CrossRef]
- Singhal, A.; Farooqi, I.S.; Cole, T.J.; O’Rahilly, S.; Fewtrell, M.; Kattenhorn, M.; Lucas, A.; Deanfield, J. Influence of leptin on arterial distensibility: A novel link between obesity and cardiovascular disease? Circulation 2002, 106, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Lin, Y.L.; Lee, C.J.; Wang, C.H.; Lai, Y.H.; Liou, H.H.; Hsu, B.G. Hyperleptinemia positively associated with central arterial stiffness in hemodialysis patients. PLoS ONE 2018, 13, e0190694. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.P.; Wang, J.H.; Chen, M.L.; Yang, C.F.; Chen, Y.C.; Hsu, B.G. Association of serum leptin levels with central arterial stiffness in coronary artery disease patients. BMC Cardiovasc. Dis. 2016, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Coleman, T.T.; Sasser, J.M.; Pittman, K.M.; Hankins, M.W.; Stec, D.E. Vascular smooth muscle-specific deletion of the leptin receptor attenuates leptin-induced alterations in vascular relaxation. Am. J. Physiol. Reg. Integ. Comp. Physiol. 2016, 310, R960–R967. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Miana, M.; Jurado-Lopez, R.; Bartolome, M.V.; Souza Neto, F.V.; Salaices, M.; Lopez-Andres, N.; Cachofeiro, V. The potential role of leptin in the vascular remodeling associated with obesity. Int. J. Obes. 2014, 38, 1565–1572. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, F.; Wang, B.J.; Chu, G.; Cao, Q.; Sun, B.G.; Dai, Q.Y. Inhibition of leptin-induced vascular extracellular matrix remodelling by adiponectin. J. Mol. Endocrinol. 2014, 53, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Mao, C.; Jia, Y.; Fu, Y.; Kong, W. Extracellular matrix dynamics in vascular remodeling. American journal of physiology. Cell Physiol. 2020, 319, C481–C499. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Lacheta, D.; Kubiak-Tomaszewska, G. Matrix metalloproteinases as biomarkers of atherosclerotic plaque instability. Int. J. Mol. Sci 2020, 21, 3946. [Google Scholar] [CrossRef]
- Schneiderman, J.; Schaefer, K.; Kolodgie, F.D.; Savion, N.; Kotev-Emeth, S.; Dardik, R.; Simon, A.J.; Halak, M.; Pariente, C.; Engelberg, I.; et al. Leptin locally synthesized in carotid atherosclerotic plaques could be associated with lesion instability and cerebral emboli. J. Am. Heart Assoc. 2012, 1, e001727. [Google Scholar] [CrossRef]
- Liu, R.; Chen, B.; Chen, J.; Lan, J. Leptin upregulates smooth muscle cell expression of MMP-9 to promote plaque destabilization by activating AP-1 via the leptin receptor/MAPK/ERK signaling pathways. Exp. Ther. Med. 2018, 16, 5327–5333. [Google Scholar] [CrossRef] [PubMed]
- Zeadin, M.; Butcher, M.; Werstuck, G.; Khan, M.; Yee, C.K.; Shaughnessy, S.G. Effect of leptin on vascular calcification in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2009, 29, 2069–2075. [Google Scholar] [CrossRef] [PubMed]
- Zeadin, M.G.; Butcher, M.K.; Shaughnessy, S.G.; Werstuck, G.H. Leptin promotes osteoblast differentiation and mineralization of primary cultures of vascular smooth muscle cells by inhibiting glycogen synthase kinase (GSK)-3beta. Biochem. Biophys. Res. Commun. 2012, 425, 924–930. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Population | Major Findings | References |
|---|---|---|
| CAD patients (± high fasting serum leptin | ↑ leptin levels ∞ ↑ cardiovascular events | [24] |
|
| [25] |
| Type 2 diabetics in varying plasma leptin quartiles | Higher leptin quartiles ∞ ↑ CAC scores | [28] |
| SLE patients vs. healthy controls |
| [29] |
| CAD patients vs. healthy controls |
| [30] |
| CAD vs. healthy controls |
| [31] |
| CAVD vs. non-CAVD patients |
| [32] |
| AMI patients vs. stable cardiac patients |
| [33] |
| Hypertensive patients (± MI) |
| [34] |
| CVD patients vs. controls (meta-analyses including 13 studies) |
| [35] |
| Young normotensive healthy adults |
| [36] |
| Animal model | Diet/Treatment | Major Findings | References |
|---|---|---|---|
| FeCl3-induced carotid artery injury in WT, ob/ob & db/db | recombinant murine leptin (0.6 µg/g, 3 weeks) ± HFD |
| [37] |
| femoral artery wire injury- induced WT, ob/ob and db/db | recombinant murine leptin (± 0.4 mg/Kg, 2 weeks) |
| [38] |
| injury-induced WT, ob/ob | normal chow; HFD |
| [39] |
| femoral artery wire injury in db/db and WT littermates |
| [40] | |
| femoral artery-induced vascular injury in WT, ob/ob, db/db | recombinant murine leptin (5 µg/g bw) adeno virus-expressing murine leptin |
| [41] |
| FeCl3-induced carotid artery injury in ApoE-/-, LDL-R-/- and WT | recombinant murine leptin (0.6 µg/g bw) |
| [42] |
| ApoE-/- | recombinant murine leptin (125 µg 1x daily for 4 weeks); western diet |
| [43] |
| ob/ob;ApoE-/- vs. ApoE-/- | atherogenic diet (16 weeks) |
| [44] |
| ob/ob;LDL-R-/- vs. LDL-R-/- | ± HFD (12 weeks) |
| [45] |
| TSP1-/-;ApoE-/- vs. ApoE-/- | recombinant murine leptin (125 µg, 1x daily for 3 weeks), ± western diet |
| [46] |
| db/db;ApoE-/-, db/+;ApoE-/- and ApoE-/- |
| [47] | |
| ob/ob;LDL-R-/- | recombinant murine leptin (0.1–3 mg/Kg bw, 12 weeks) |
| [48] |
| Ins2+/Akita;ApoE-/- vs. Ins2+/+;ApoE-/- | recombinant murine leptin (0.4 µg/g bw, 1x daily for 12 weeks) |
| [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, P.; Khanal, S. Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. Int. J. Mol. Sci. 2021, 22, 5446. https://doi.org/10.3390/ijms22115446
Raman P, Khanal S. Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. International Journal of Molecular Sciences. 2021; 22(11):5446. https://doi.org/10.3390/ijms22115446
Chicago/Turabian StyleRaman, Priya, and Saugat Khanal. 2021. "Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells" International Journal of Molecular Sciences 22, no. 11: 5446. https://doi.org/10.3390/ijms22115446
APA StyleRaman, P., & Khanal, S. (2021). Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. International Journal of Molecular Sciences, 22(11), 5446. https://doi.org/10.3390/ijms22115446