
RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. RABL6A Expression Inversely Correlates with Markers of Cellular Senescence in Primary NHSCs
2.2. Loss of RABL6A Causes Premature Stress-Induced Senescence in Primary NHSCs
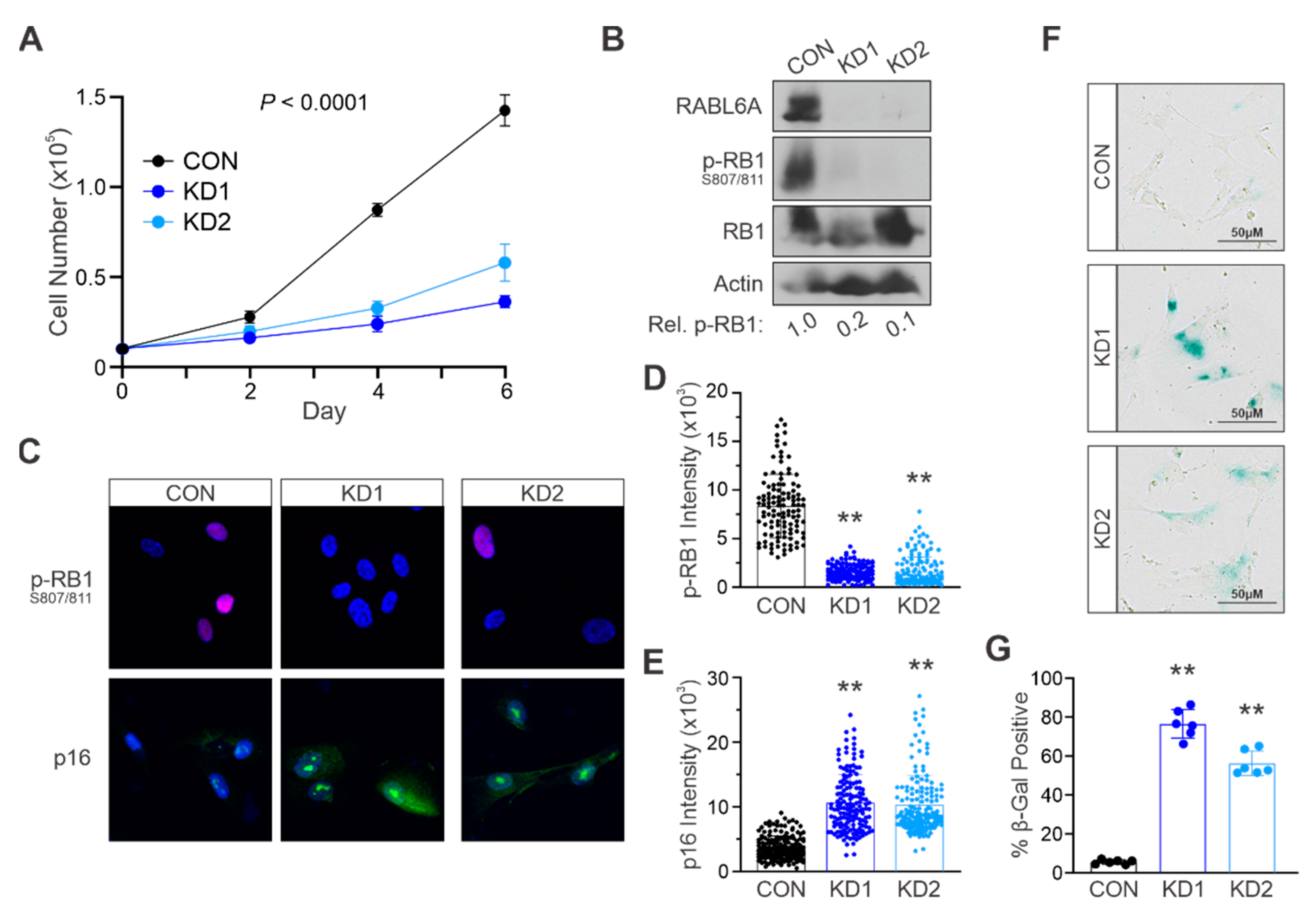
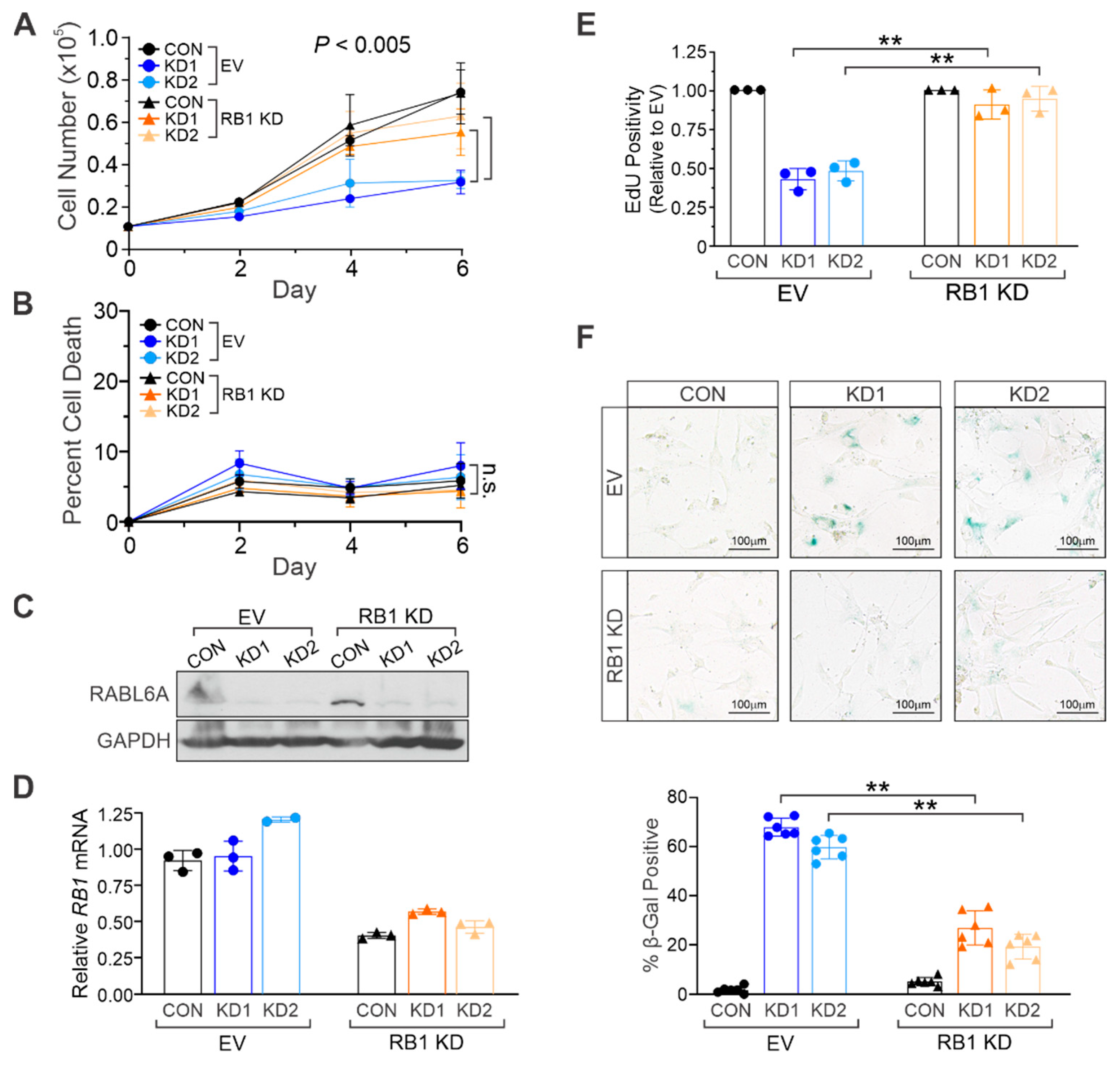
2.3. RABL6A Promotes NHSC Proliferation and Evasion of Senescence via RB1 Inactivation
2.4. RABL6A Loss in Mouse Embryonic Fibroblasts Reduces Proliferative Capacity and Causes Premature Senescence
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Interference, virus Production, and Infection
4.3. Cell Proliferation and Senescence Assays
4.4. Antibodies
4.5. Western Blotting
4.6. Immunofluorescence
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Biology of Schwann cells. Handb. Clin. Neurol. 2013, 115, 55–79. [Google Scholar] [PubMed]
- Carroll, S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012, 123, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Quelle, D.; Nteeba, J.; Darbro, B. The INK4a/ARF locus. Encycl. Cell Biol. 2016, 3, 447–457. [Google Scholar]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Toussaint, O.; Royer, V.; Salmon, M.; Remacle, J. Stress-induced premature senescence and tissue ageing. Biochem. Pharmacol. 2002, 64, 1007–1009. [Google Scholar] [CrossRef]
- Chen, Q.M.; Prowse, K.R.; Tu, V.C.; Purdom, S.; Linskens, M.H. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp. Cell Res. 2001, 265, 294–303. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Gomis-Coloma, C.; Morenilla-Palao, C.; Peiro, G.; Serra, E.; Serrano, M.; Cabedo, H. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain 2013, 136 Pt 7, 2262–2278. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997–3011. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.P.; Askeland, R.W.; Zhang, X.; Reed, S.M.; Tompkins, V.S.; Hagen, J.; McDowell, B.D.; Button, A.; Smith, B.J.; Weydert, J.A.; et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes Cancer 2013, 4, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, J.; Lui, K.; Sheikh, M.S.; Huang, Y. Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J. Biol. Chem. 2009, 284, 18129–18142. [Google Scholar] [CrossRef]
- Umesalma, S.; Kaemmer, C.A.; Kohlmeyer, J.L.; Letney, B.; Schab, A.M.; Reilly, J.A.; Sheehy, R.M.; Hagen, J.; Tiwari, N.; Zhan, F.; et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J. Clin. Investig. 2019, 129, 1641–1653. [Google Scholar] [CrossRef]
- Lui, K.; An, J.; Montalbano, J.; Shi, J.; Corcoran, C.; He, Q.; Sun, H.; Sheikh, M.S.; Huang, Y. Negative regulation of p53 by Ras superfamily protein RBEL1A. J. Cell Sci. 2013, 126 Pt 11, 2436–2445. [Google Scholar] [CrossRef]
- Hagen, J.; Muniz, V.P.; Falls, K.C.; Reed, S.M.; Taghiyev, A.F.; Quelle, F.W.; Gourronc, F.A.; Klingelhutz, A.J.; Major, H.J.; Askeland, R.W.; et al. RABL6A promotes G1-S phase progression and pancreatic neuroendocrine tumor cell proliferation in an Rb1-dependent manner. Cancer Res. 2014, 74, 6661–6670. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Ji, F.; Sun, J.; Xie, Y.; Xu, Y.; Yue, H. RBEL1 is required for osteosarcoma cell proliferation via inhibiting retinoblastoma 1. Mol. Med. Rep. 2016, 13, 1275–1280. [Google Scholar] [CrossRef]
- Maharjan, C.K.; Kaemmer, C.A.; Muniz, V.P.; Bauchle, C.; Mott, S.L.; Zamba, K.D.; Breheny, P.; Leidinger, M.R.; Darbro, B.W.; Stephens, S.; et al. RABL6A promotes pancreatic neuroendocrine tumor angiogenesis and progression in vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 7429697. [Google Scholar] [CrossRef]
- Reilly, K.M.; Kim, A.; Blakely, J.; Ferner, R.E.; Gutmann, D.H.; Legius, E.; Miettinen, M.M.; Randall, R.L.; Ratner, N.; Jumbé, N.L.; et al. Neurofibromatosis Type 1-Associated MPNST State of the Science: Outlining a Research Agenda for the Future. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Gordon, D.J.; Tanas, M.R.; Dodd, R.D.; Monga, V.; Darbro, B.W.; Quelle, D.E. Combination therapies for MPNSTs targeting RABL6A-RB1 signaling. Oncotarget 2021, 12, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef]
- Tompkins, V.; Hagen, J.; Zediak, V.P.; Quelle, D.E. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle. 2006, 5, 641–646. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohlmeyer, J.L.; Kaemmer, C.A.; Umesalma, S.; Gourronc, F.A.; Klingelhutz, A.J.; Quelle, D.E. RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 5367. https://doi.org/10.3390/ijms22105367
Kohlmeyer JL, Kaemmer CA, Umesalma S, Gourronc FA, Klingelhutz AJ, Quelle DE. RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner. International Journal of Molecular Sciences. 2021; 22(10):5367. https://doi.org/10.3390/ijms22105367
Chicago/Turabian StyleKohlmeyer, Jordan L., Courtney A. Kaemmer, Shaikamjad Umesalma, Francoise A. Gourronc, Aloysius J. Klingelhutz, and Dawn E. Quelle. 2021. "RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner" International Journal of Molecular Sciences 22, no. 10: 5367. https://doi.org/10.3390/ijms22105367
APA StyleKohlmeyer, J. L., Kaemmer, C. A., Umesalma, S., Gourronc, F. A., Klingelhutz, A. J., & Quelle, D. E. (2021). RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner. International Journal of Molecular Sciences, 22(10), 5367. https://doi.org/10.3390/ijms22105367