
The Detection and Bioinformatic Analysis of Alternative 3′ UTR Isoforms as Potential Cancer Biomarkers

 ,
,  , and
, and
Abstract
1. Introduction
2. Implications of Alternative Polyadenylation
3. Next-Generation Sequencing Based Techniques for Characterisation of APA
3.1. 3 focused RNA-seq Methods for APA Characterisation
3.2. Single-Cell Methods for mRNA 3 End Sequencing
4. Bioinformatic Methods for Detection of Poly(A) Sites
4.1. Databases for 3 UTR and APA Storage and Retrieval
4.2. Bioinformatic Methods for APA Detection and Quantification
4.2.1. APA Detection in RNA-seq Data Based on Prior APA Information
4.2.2. de novo APA Detection in RNA-seq Data
4.2.3. de novo APA Detection in 3 Focused Data
4.2.4. APA Detection in 3 Tag-Based Single-Cell RNA-seq Data
5. The Repertoire of Cancer Biomarkers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sweet, T.J.; Licatalosi, D.D. 3’end formation and regulation of eukaryotic mRNAs. Methods Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. 3’end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008, 27. [Google Scholar] [CrossRef]
- Ozsolak, F.; Kapranov, P.; Foissac, S.; Kim, S.W.; Fishilevich, E.; Monaghan, A.P.; John, B.; Milos, P.M. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 2010, 143. [Google Scholar] [CrossRef]
- Jan, C.H.; Friedman, R.C.; Ruby, J.G.; Bartel, D.P. Formation, regulation and evolution of Caenorhabditis elegans 3’UTRs. Nature 2011, 469, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Shepard, P.J.; Choi, E.A.; Lu, J.; Flanagan, L.A.; Hertel, K.J.; Shi, Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 2011, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark. Res. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; MacDonald, C.C.; Graber, J.H. Systematic variation in mRNA 3’-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, H.K.; Alcott, C.E.; Ji, P.; Wagner, E.J.; Zoghbi, H.Y.; Liu, Z. PolyA-miner: Accurate assessment of differential alternative poly-adenylation from 3′Seq data using vector projections and non-negative matrix factorization. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.C.H.; Blencowe, B.J.; Morris, Q. QAPA: A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.C.; Zheng, D.; Baljinnyam, E.; Sun, F.; Ogami, K.; Yeung, P.L.; Hoque, M.; Lu, C.W.; Manley, J.L.; Tian, B. Widespread transcript shortening through alternative polyadenylation in secretory cell differentiation. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Mayr, C. Alternative 3’UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef]
- Mayr, C. Evolution and Biological Roles of Alternative 3’UTRs. Trends Cell Biol. 2016, 26. [Google Scholar] [CrossRef]
- Millevoi, S.; Vagner, S. Molecular mechanisms of eukaryotic pre-mRNA 3’ end processing regulation. Nucleic Acids Res. 2010, 38, 2757–2774. [Google Scholar] [CrossRef]
- Giammartino, D.C.D.; Nishida, K.; Manley, J.L. Mechanisms and Consequences of Alternative Polyadenylation. Mol. Cell 2011, 43. [Google Scholar] [CrossRef]
- Chen, W.; Jia, Q.; Song, Y.; Fu, H.; Wei, G.; Ni, T. Alternative Polyadenylation: Methods, Findings, and Impacts. Genom. Proteom. Bioinform. 2017, 15, 287–300. [Google Scholar] [CrossRef]
- Turner, R.E.; Pattison, A.D.; Beilharz, T.H. Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell Dev. Biol. 2018, 75. [Google Scholar] [CrossRef]
- Rogers, J.; Early, P.; Carter, C.; Calame, K.; Bond, M.; Hood, L.; Wall, R. Two mRNAs with different 3’ends encode membrane-bound and secreted forms of immunoglobulin μ chain. Cell 1980, 20. [Google Scholar] [CrossRef]
- Setzer, D.R.; McGrogan, M.; Nunberg, J.H.; Schimke, R.T. Size heterogeneity in the 3′end of dihydrofolate reductase messenger RNAs in mouse cells. Cell 1980, 22. [Google Scholar] [CrossRef]
- Chatterjee, S.; Pal, J.K. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.B.; Oyken, M.; Tuncer, T.; Can, T.; Erson-Bensan, A.E. 3’UTR shortening and EGF signaling: Implications for breast cancer. Hum. Mol. Genet. 2015, 24, 6910–6920. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Tian, B. Reprogramming of 3’ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 2009, 4, e8419. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef]
- Thivierge, C.; Tseng, H.W.; Mayya, V.K.; Lussier, C.; Gravel, S.P.; Duchaine, T.F. Alternative polyadenylation confers Pten mRNAs stability and resistance to microRNAs. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef]
- Hong, W.; Ruan, H.; Zhang, Z.; Ye, Y.; Liu, Y.; Li, S.; Jing, Y.; Zhang, H.; Diao, L.; Liang, H.; et al. APAatlas: Decoding alternative polyadenylation across human tissues. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef]
- Zhang, H.; Lee, J.Y.; Tian, B. Biased alternative polyadenylation in human tissues. Genome. Biol. 2005, 6, R100. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Lianoglou, S.; Garg, V.; Yang, J.L.; Leslie, C.S.; Mayr, C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013, 27, 2380–2396. [Google Scholar] [CrossRef]
- Lee, S.H.; Singh, I.; Tisdale, S.; Abdel-Wahab, O.; Leslie, C.S.; Mayr, C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 2018, 561, 127–131. [Google Scholar] [CrossRef]
- Singh, I.; Lee, S.H.; Sperling, A.S.; Samur, M.K.; Tai, Y.T.; Fulciniti, M.; Munshi, N.C.; Mayr, C.; Leslie, C.S. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3’ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef]
- Singh, P.; Alley, T.L.; Wright, S.M.; Kamdar, S.; Schott, W.; Wilpan, R.Y.; Mills, K.D.; Graber, J.H. Global changes in processing of mRNA 3’ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009, 69, 9422–9430. [Google Scholar] [CrossRef]
- Flavell, S.W.; Kim, T.K.; Gray, J.M.; Harmin, D.A.; Hemberg, M.; Hong, E.J.; Markenscoff-Papadimitriou, E.; Bear, D.M.; Greenberg, M.E. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 2008, 60, 1022–1038. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, X.; Tian, B. 3’READS+, a sensitive and accurate method for 3’ end sequencing of polyadenylated RNA. RNA 2016, 22, 1631–1639. [Google Scholar] [CrossRef]
- Xue, Z.; Warren, R.L.; Gibb, E.A.; MacMillan, D.; Wong, J.; Chiu, R.; Hammond, S.A.; Yang, C.; Nip, K.M.; Ennis, C.A.; et al. Recurrent tumor-specific regulation of alternative polyadenylation of cancer-related genes. BMC Genom. 2018, 19. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019, 20. [Google Scholar] [CrossRef]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; Haaften, G.V.; Kühn, U.; Menzies, F.M.; Vrielink, J.A.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149. [Google Scholar] [CrossRef]
- Gruber, A.J.; Schmidt, R.; Ghosh, S.; Martin, G.; Gruber, A.R.; van Nimwegen, E.; Zavolan, M. Discovery of physiological and cancer-related regulators of 3’ UTR processing with KAPAC. Genome Biol. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, Y.; Seipelt, R.L.; Peterson, M.L.; Manley, J.L. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 1996, 87, 941–952. [Google Scholar] [CrossRef]
- Naveed, A.; Cooper, J.A.; Li, R.; Hubbard, A.; Chen, J.; Liu, T.; Wilton, S.D.; Fletcher, S.; Fox, A.H. NEAT1 polyA-modulating antisense oligonucleotides reveal opposing functions for both long non-coding RNA isoforms in neuroblastoma. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Edwalds-Gilbert, G.; Veraldi, K.L.; Milcarek, C. Alternative poly(A) site selection in complex transcription units: Means to an end? Nucleic Acids Res. 1997, 25. [Google Scholar] [CrossRef]
- Gautheret, D.; Poirot, O.; Lopez, F.; Audic, S.; Claverie, J.M. Alternate polyadenylation in human mRNAs: A large-scale analysis by EST clustering. Genome Res. 1998, 8. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10. [Google Scholar] [CrossRef]
- Smyth, G.K.; Ritchie, M.E.; Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2018, 5. [Google Scholar] [CrossRef]
- Ma, F.; Fuqua, B.K.; Hasin, Y.; Yukhtman, C.; Vulpe, C.D.; Lusis, A.J.; Pellegrini, M. A comparison between whole transcript and 3’ RNA sequencing methods using Kapa and Lexogen library preparation methods 06 Biological Sciences 0604 Genetics. BMC Genom. 2019, 20. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. Transcriptome profiling using single-molecule direct RNA sequencing. Methods Mol. Biol. 2011, 733. [Google Scholar] [CrossRef]
- Scotto-Lavino, E.; Du, G.; Frohman, M.A. 3’End cDNA amplification using classic RACE. Nat. Protoc. 2007, 1. [Google Scholar] [CrossRef]
- Liu, Y.; Nie, H.; Liu, H.; Lu, F. Poly(A) inclusive RNA isoform sequencing (PAIso-seq) reveals wide-spread non-adenosine residues within RNA poly(A) tails. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Krause, M.; Niazi, A.M.; Labun, K.; Cleuren, Y.N.T.; Müller, F.S.; Valen, E. TailFindR: Alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA 2019, 25. [Google Scholar] [CrossRef]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly(A) tail length and 3’ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Lim, J.; Lee, M.; Son, A.; Chang, H.; Kim, V.N. MTAIL-seq reveals dynamic poly(A) tail regulation in oocyte-to-embryo development. Genes Dev. 2016, 30. [Google Scholar] [CrossRef]
- Harrison, P.F.; Powell, D.R.; Clancy, J.L.; Preiss, T.; Boag, P.R.; Traven, A.; Seemann, T.; Beilharz, T.H. PAT-seq: A method to study the integration of 3’-UTR dynamics with gene expression in the eukaryotic transcriptome. RNA 2015, 21, 1502–1510. [Google Scholar] [CrossRef]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.; Bartel, D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 2014, 508, 66–71. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, Y.; Cheng, C.; Wang, W.; Zhou, Z.; Rang, W.; Yu, H.; Wei, Y.; Wu, Q.; Zhang, Y. Poly(A)-seq: A method for direct sequencing and analysis of the transcriptomic poly(A)-tails. PLoS ONE 2020, 15, e0234696. [Google Scholar] [CrossRef]
- Woo, Y.M.; Kwak, Y.; Namkoong, S.; Kristjánsdóttir, K.; Lee, S.H.; Lee, J.H.; Kwak, H. TED-Seq Identifies the Dynamics of Poly(A) Length during ER Stress. Cell Rep. 2018, 24. [Google Scholar] [CrossRef]
- Spies, N.; Burge, C.B.; Bartel, D.P. 3’ UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome. Res. 2013, 23, 2078–2090. [Google Scholar] [CrossRef]
- Mata, J. Genome-wide mapping of polyadenylation sites in fission yeast reveals widespread alternative polyadenylation. RNA Biol. 2013, 10, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, S.; Pelechano, V.; Jarvelin, A.I.; Tekkedil, M.M.; Anders, S.; Benes, V.; Steinmetz, L.M. An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res. 2013, 41, e65. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, Y.; Li, Y.; Li, J.; Rao, X.; Chen, C.; Xu, A. Differential genome-wide profiling of tandem 3’ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome. Res. 2011, 21, 741–747. [Google Scholar] [CrossRef]
- Fu, Y.; Ge, Y.; Sun, Y.; Liang, J.; Wan, L.; Wu, X.; Xu, A. IVT-SAPAS: Low-Input and Rapid Method for Sequencing Alternative Polyadenylation Sites. PLoS ONE 2015, 10, e0145477. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.W.; Park, C.Y.; Goodarzi, H.; Fak, J.J.; Mele, A.; Moore, M.J.; Saito, Y.; Darnell, R.B. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 2016, 15, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Müller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Moll, P.; Ante, M.; Seitz, A.; Reda, T. QuantSeq 3’mRNA sequencing for RNA quantification. Nat. Methods 2014, 11, i–iii. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, H.R.; Huang, J.; Jin, G.; Fu, X.D. Multiplex analysis of polyA-linked sequences (MAPS): An RNA-Seq strategy to profile poly(A+) RNA. Methods Mol. Biol. 2014, 1125. [Google Scholar] [CrossRef]
- Pallares, L.F.; Picard, S.; Ayroles, J.F. TM3’seq: A tagmentation-mediated 3’sequencing approach for improving scalability of RNAseq experiments. G3 Genes Genomes Genetics 2020, 10, 143–150. [Google Scholar] [CrossRef]
- Routh, A.; Ji, P.; Jaworski, E.; Xia, Z.; Li, W.; Wagner, E.J. Poly(A)-ClickSeq: Click-chemistry for next-generation 3-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017, 45, e112. [Google Scholar] [CrossRef]
- Welch, J.D.; Slevin, M.K.; Tatomer, D.C.; Duronio, R.J.; Prins, J.F.; Marzluff, W.F. EnD-Seq and AppEnD: Sequencing 3’ ends to identify nontemplated tails and degradation intermediates. RNA 2015, 21. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yeh, I.; Park, J.Y.; Tian, B. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome. Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Hwang, H.W.; Darnell, R.B. Comprehensive Identification of mRNA Polyadenylation Sites by PAPERCLIP. Methods Mol. Biol. 2017, 1648, 79–93. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef]
- Camp, J.G.; Wollny, D.; Treutlein, B. Single-cell genomics to guide human stem cell and tissue engineering. Nat. Methods 2018, 15, 661–667. [Google Scholar] [CrossRef]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome. Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Islam, S.; Zeisel, A.; Joost, S.; Manno, G.L.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161. [Google Scholar] [CrossRef]
- Patrick, R.; Humphreys, D.T.; Janbandhu, V.; Oshlack, A.; Ho, J.W.; Harvey, R.P.; Lo, K.K. Sierra: Discovery of differential transcript usage from polyA-captured single-cell RNA-seq data. Genome. Biol. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, T.; Ye, C.; Ye, W.; Ji, G. scAPAtrap: Identification and quantification of alternative polyadenylation sites from single-cell RNA-seq data. Briefings Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Chung, W.; Eum, H.H.; Lee, H.O.; Park, W.Y. Alternative polyadenylation of single cells delineates cell types and serves as a prognostic marker in early stage breast cancer. PLoS ONE 2019, 14, e0217196. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lee, D.; Bang, D.; Lee, J.H. MAPS-seq: Magnetic bead-assisted parallel single-cell gene expression profiling. Exp. Mol. Med. 2020, 52, 804–814. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2. [Google Scholar] [CrossRef]
- Yanai, I.; Hashimshony, T. CEL-Seq2—Single-cell RNA sequencing by multiplexed linear amplification. Methods Mol. Biol. 2019, 1979. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Kenigsberg, E.; Jaitin, D.A.; David, E.; Paul, F.; Tanay, A.; Amit, I. MARS-seq2.0: An experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 2019, 14. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161. [Google Scholar] [CrossRef]
- Soumillon, M.; Cacchiarelli, D.; Semrau, S.; van Oudenaarden, A.; Mikkelsen, T. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. bioRxiv 2014. [Google Scholar] [CrossRef]
- Velten, L.; Anders, S.; Pekowska, A.; Jarvelin, A.I.; Huber, W.; Pelechano, V.; Steinmetz, L.M. Single-cell polyadenylation site mapping reveals 3’ isoform choice variability. Mol. Syst. Biol. 2015, 11, 812. [Google Scholar] [CrossRef]
- Pesole, G.; Liuni, S.; Grillo, G.; Saccone, C. UTRdb: A specialized database of 5’- and 3’-untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 1998, 26, 192–195. [Google Scholar] [CrossRef][Green Version]
- Wang, R.; Nambiar, R.; Zheng, D.; Tian, B. PolyA-DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef]
- Brockman, J.M.; Singh, P.; Liu, D.; Quinlan, S.; Salisbury, J.; Graber, J.H. PACdb: PolyA Cleavage Site and 3’-UTR Database. Bioinformatics 2005, 21, 3691–3693. [Google Scholar] [CrossRef][Green Version]
- You, L.; Wu, J.; Feng, Y.; Fu, Y.; Guo, Y.; Long, L.; Zhang, H.; Luan, Y.; Tian, P.; Chen, L.; et al. APASdb: A database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 2015, 43, D59–D67. [Google Scholar] [CrossRef]
- Muller, S.; Rycak, L.; Afonso-Grunz, F.; Winter, P.; Zawada, A.M.; Damrath, E.; Scheider, J.; Schmah, J.; Koch, I.; Kahl, G.; et al. APADB: A database for alternative polyadenylation and microRNA regulation events. Database 2014, 2014. [Google Scholar] [CrossRef]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3’ end sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef]
- Feng, X.; Li, L.; Wagner, E.J.; Li, W. TC3A: The Cancer 3’UTR Atlas. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome. Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Wspolczesna Onkol. 2015, 1A. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, J.; Recce, M.; Tian, B. PolyA_DB: A database for mammalian mRNA polyadenylation. Nucleic Acids Res. 2005, 33, D116–D120. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7. [Google Scholar] [CrossRef]
- Grassi, E.; Mariella, E.; Lembo, A.; Molineris, I.; Provero, P. Roar: Detecting alternative polyadenylation with standard mRNA sequencing libraries. BMC Bioinform. 2016, 17, 423. [Google Scholar] [CrossRef]
- Pera, L.L.; Mazzapioda, M.; Tramontano, A. 3USS: A web server for detecting alternative 3’UTRs from RNA-seq experiments. Bioinformatics 2015, 31, 1845–1847. [Google Scholar] [CrossRef]
- Fahmi, N.A.; Chang, J.W.; Nassereddeen, H.; Ahmed, K.T.; Fan, D.; Yong, J.; Zhang, W. APA-Scan: Detection and Visualization of 3’-UTR APA with RNA-seq and 3’-end-seq Data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guvenek, A.; Tian, B. Analysis of alternative cleavage and polyadenylation in mature and differentiating neurons using RNA-seq data. Quant. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Wang, R.; Tian, B. APAlyzer: A bioinformatics package for analysis of alternative polyadenylation isoforms. Bioinformatics 2020, 36. [Google Scholar] [CrossRef]
- Arefeen, A.; Liu, J.; Xiao, X.; Jiang, T. TAPAS: Tool for alternative polyadenylation site analysis. Bioinformatics 2018, 34, 2521–2529. [Google Scholar] [CrossRef]
- Kim, M.; You, B.H.; Nam, J.W. Global estimation of the 3’ untranslated region landscape using RNA sequencing. Methods 2015, 83, 111–117. [Google Scholar] [CrossRef]
- Shenker, S.; Miura, P.; Sanfilippo, P.; Lai, E.C. IsoSCM: Improved and alternative 3’ UTR annotation using multiple change-point inference. RNA 2015, 21, 14–27. [Google Scholar] [CrossRef]
- Bicknell, A.A.; Cenik, C.; Chua, H.N.; Roth, F.P.; Moore, M.J. Introns in UTRs: Why we should stop ignoring them. BioEssays 2012, 34. [Google Scholar] [CrossRef]
- Barrett, L.W.; Fletcher, S.; Wilton, S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012, 69. [Google Scholar] [CrossRef]
- Ye, C.; Long, Y.; Ji, G.; Li, Q.Q.; Wu, X. APAtrap: Identification and quantification of alternative polyadenylation sites from RNA-seq data. Bioinformatics 2018, 34. [Google Scholar] [CrossRef]
- Wang, W.; Wei, Z.; Li, H. A change-point model for identifying 3’UTR switching by next-generation RNA sequencing. Bioinformatics 2014, 30. [Google Scholar] [CrossRef]
- Harrison, P.F. Tools for Matrices with Precision Weights, Test and Explore Weighted or Sparse Data. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/weitrix.html (accessed on 8 April 2021). [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef]
- Harrison, P.F.; Pattison, A.D.; Powell, D.R.; Beilharz, T.H. Topconfects: A package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Ensembl insights: How are UTRs annotated? Ensembl Blog. Available online: https://www.ensembl.info/ (accessed on 8 April 2021).
- Cass, A.A.; Xiao, X. mountainClimber Identifies Alternative Transcription Start and Polyadenylation Sites in RNA-Seq. Cell Syst. 2019, 9, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Liu, T.; Fu, H.; Ye, C.; Ji, G.; Wu, X. movAPA: Modeling and visualization of dynamics of alternative polyadenylation across biological samples. Bioinformatics 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ye, W.; Ye, L.; Fu, H.; Ye, C.; Xiao, X.; Ji, Y.; Lin, W.; Ji, G.; Wu, X. PlantAPAdb: A comprehensive database for alternative polyadenylation sites in plants. Plant Physiol. 2020, 182. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.J.V.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28. [Google Scholar] [CrossRef]
- Shulman, E.D.; Elkon, R. Cell-type-specific analysis of alternative polyadenylation using single-cell transcriptomics data. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22. [Google Scholar] [CrossRef]
- Ye, C.; Zhou, Q.; Wu, X.; Yu, C.; Ji, G.; Saban, D.R.; Li, Q.Q. ScDAPA: Detection and visualization of dynamic alternative polyadenylation from single cell RNA-seq data. Bioinformatics 2020, 36. [Google Scholar] [CrossRef]
- Masamha, C.P.; Wagner, E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 2018, 39, 2–10. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Xiang, Y.; Ye, Y.; Lou, Y.; Yang, Y.; Cai, C.; Zhang, Z.; Mills, T.; Chen, N.Y.; Kim, Y.; Ozguc, F.M.; et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018, 110, 379–389. [Google Scholar] [CrossRef]
- Wang, L.; Hu, X.; Wang, P.; Shao, Z.M. The 3’UTR signature defines a highly metastatic subgroup of triple-negative breast cancer. Oncotarget 2016, 7, 59834–59844. [Google Scholar] [CrossRef]
- Gillen, A.E.; Brechbuhl, H.M.; Yamamoto, T.M.; Kline, E.; Pillai, M.M.; Hesselberth, J.R.; Kabos, P. Alternative Polyadenylation of PRELID1 Regulates Mitochondrial ROS Signaling and Cancer Outcomes. Mol. Cancer Res. 2017, 15, 1741–1751. [Google Scholar] [CrossRef]
- Schwab, M. MammaPrint Test. Encycl. Cancer 2015. [Google Scholar] [CrossRef]
- Jensen, M.B.; Lænkholm, A.V.; Nielsen, T.O.; Eriksen, J.O.; Wehn, P.; Hood, T.; Ram, N.; Buckingham, W.; Ferree, S.; Ejlertsen, B. The Prosigna gene expression assay and responsiveness to adjuvant cyclophosphamide-based chemotherapy in premenopausal high-risk patients with breast cancer. Breast Cancer Res. 2018, 20. [Google Scholar] [CrossRef]
- Andres, S.F.; Williams, K.N.; Plesset, J.B.; Headd, J.J.; Mizuno, R.; Chatterji, P.; Lento, A.A.; Klein-Szanto, A.J.; Mick, R.; Hamilton, K.E.; et al. IMP1 3’UTR shortening enhances metastatic burden in colorectal cancer. Carcinogenesis 2019, 40. [Google Scholar] [CrossRef]
- Triple-Negative Breast Cancer: Overview, Treatment, and More. Available online: Breastcancer.org (accessed on 8 April 2021).
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef]
- Ogorodnikov, A.; Levin, M.; Tattikota, S.; Tokalov, S.; Hoque, M.; Scherzinger, D.; Marini, F.; Poetsch, A.; Binder, H.; Macher-Göppinger, S.; et al. Transcriptome 3’ end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018, 9, 5331. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
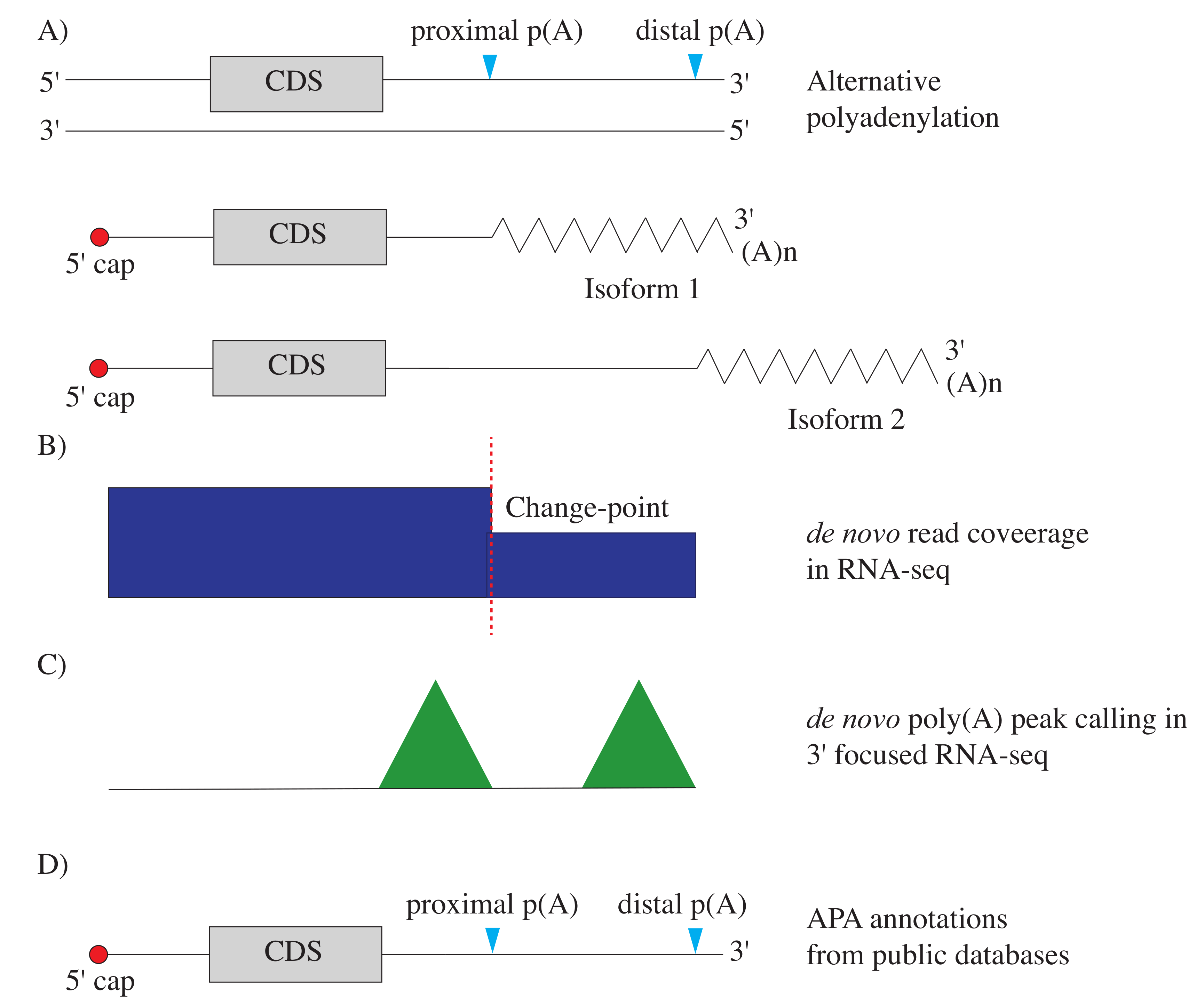{kind=link}
| Name | Key Points | Typical Input | Sequence Target |
|---|---|---|---|
| PAIso-seq [52] | PacBio based method to capture poly(A) site, length, splicing, expression, PacBio is costly for the read coverage obtained, Low coverage | 100 ng total RNA | Full length mRNA, Poly(A) tail included |
| Oxford Nanopore- Direct RNA sequencing [53] | The Nanopore instrument is capable of full-length direct RNA seq, tail lengths can also be extracted. Low coverage | 500 ng poly(A)+ selected RNA | Full length mRNA, Poly(A) tail included |
| TAIL-seq [54] | rRNA depletion and 3 adaptor ligation, asymmetric paired end sequencing to determine tail length | ∼100 g total RNA | Poly(A) tail length, Poly(A) site |
| mTAIL-seq [55] | 3 oligo(dT) splinted ligation approach to TAIL-seq, reduced input RNA required. Paired-end sequencing. | 1–5 g total RNA | Poly(A) tail length, Poly(A) site |
| PAT-seq [56] | Single end read approach, 3 tagging by oligo templated RNA end extension | 1 g total RNA | Poly(A) tail length, Poly(A) site |
| PAL-seq [57] | Requires non-standard use of an Illumina instrument for tail length measurement by biotinylated dTTP incorporation. 3 end capture by splinted ligation | 1–50 g total RNA | Poly(A) tail length, Poly(A) site |
| Poly(A) seq [58] | Poly(A)+ RNA is captured with oligo(dT) conjugated magnetic beads, then 3 adaptors ligated 300 bp single end read. Samples sequenced on the Illumina NextSeq 500, 2 colour sequencing instrument | 5.1 g total RNA | Poly(A) tail length, Poly(A) site |
| TED-Seq [59] | 3 adaptor ligation to Poly(A)+ RNA. Tail length is inferred from the size of the templated sequence after precise library size selection | 100 ng poly(A)+ RNA | Poly(A) tail length, Poly(A) site |
| 3P-seq [4] | Poly(A) tail removed by RNase H. Sequenced from the 3 end to determine site usage, adaptor addition by ligation to avoid internal priming | 30 g total RNA | Poly(A) site |
| 2P-seq [60] | Poly(A) site detection by anchored oligo(dT) priming, sequencing from start of poly(A) tail in reverse | 15 g total RNA | Poly(A) site |
| 3-seq [30] | Poly(A) site detection by anchored oligo(dT) priming. Unique approach to fragmentation by rate limited nick translation of double stranded cDNA | 2 g DNase treated RNA | Poly(A) site |
| 3READS+ [37] | Poly(A) tail is trimmed by RNase H, 3 adapter ligation | 0.1–15 g total RNA | Poly(A) site |
| 3PC [61] | Anchored oligo(dT) primer to detect poly(A) site, 5 adaptor addition by circular ligation | 100 g total RNA | Poly(A) site |
| 3T-fill [62] | Anchored oligo(dT) primer to detect poly(A) site, sequenced from 3 end. 3T-fill reaction - dA homopolymer region at 3 end filled with dTTPs on Illumina cBot cluster station before sequencing | 0.5–10 g total RNA | Poly(A) site |
| SAPAS [63] | Anchored oligo(dT) primer to detect poly(A) site, 5 adaptor addition by template switching | 10 g total RNA | Poly(A) site |
| PAS-seq [5] | Anchored oligo(dT) primer to detect poly(A) site, template switching 5 adaptor addition | 0.5–1 g poly(A)+ selected RNA | Poly(A) site |
| IVT-SAPAS [64] | in vitro transcription based amplification of cDNA for low input samples, poly(A) site detection by anchored oligo(dT) annealing | 200 ng total RNA | Poly(A) site |
| PAPERCLIP [65] | RNA crosslinked, partially digested, and 3 ends immunoprecipitated via Poly(A) Binding protein, addresses internal priming issues, uses anchored oligo(dT) annealing for end detection | NA, starting material is tissue/cells | Poly(A) site |
| MACE [66] | GenXPro commercial kit, barcodes transcripts with UMIs to deal with PCR duplication | 0.05 ng total RNA | Poly(A) site |
| Quant-Seq [67] | Lexogen commercial kit, oligo(dT) annealing to detect 3 ends, random forward priming of 2nd strand cDNA adds 5 adaptor | 0.5–500 ng total RNA | Poly(A) site |
| MAPS [68] | 3 end detection by anchored oligo(dT) priming, 5 adaptor addition by random forward priming of 2nd stand cDNA | 1 g total RNA | Poly(A) site |
| TM3seq [69] | Fragmentation and 5 adaptor addition combined in a single step. 3 end detected via annealing of oligo(dT) primer | 200 ng total RNA | Poly(A) site |
| PAC-seq [70] | Click-chemistry approach to fragmentation and 5 adaptor addition via reverse transcription termination by 3-azido-nucleotides. 3 end detected by oligo(dT) annealing | 0.125–4 g total RNA | Poly(A) site |
| EnD-Seq [71] | Targeted sequencing approach to 3 end detection, 3 adaptor ligation to total RNA, gene specific multiplex PCR of cDNA | 1.5 g total RNA | Poly(A) site, non-Poly(A) 3 ends |
| Name | Overview | Scale |
|---|---|---|
| CEL-seq [86] | 3 ends enriched by anchored oligo(dT) annealing including T7 promotor. cDNA amplified by in vitro transcription (IVT), amplified RNA fragmented and ligated to adaptor. | Manually isolated single cells |
| CEL-seq2 [87,88] | Application of CEL-seq to high throughput sequencing, UMI’s added to reverse transcription oligo | Automated microfluidic sorting via Fluidigm C1 into wells |
| MARS-seq 2.0 [89] | 3 end enrichment by anchored oligo(dT) annealing, included T7 promotor. cDNA amplified via IVT | 384-well plate, FACS sorting |
| InDrop [80] | Application of CEL-seq to droplet-based sequencing for higher throughput | Droplet sequencing, inDrop system, 1CellBio |
| Drop-seq [90] | 3 enrichment by oligo(dT) annealing RT, full length cDNA 5 labelled by template switching, oligo’s with common barcode bound to beads, and separated into droplets. library prepared by Illlumina Nextera XT DNA library prep kit | Droplet sequencing, custom instrument |
| 10X Chromium [85] | 3 enrichment by anchored oligo(dT) annealing, oligo’s with common barcode bound to beads, and separated into droplets; library preparation with commercial kit GemCode Single-Cell 3 Gel Bead and library kit (now Chromium 10X) | Droplet sequencing, 10X genomics instrument |
| SCRB-seq [91] | 3 enrichment by anchored oligo(dT) primer, template switching reaction for full length cDNA, library prepared by Illlumina Nextera XT DNA library prep kit | 384-well plate, FACS sorting |
| MAPS-seq [84] | 3 ends enriched by biotinylated oligo(dT) annealing, RNA transcripts pulled down and samples pooled together using magnetic beads before RT. Full length cDNA 5 adaptor added via template switching, library prepared by Illlumina Nextera XT DNA library prep kit | 96-well plate, FACS sorting |
| BATSeq [92] | Method specifically developed to detect APA. 3 ends enriched by oligo(dT) annealing. 2nd strand cDNA IVT amplified | FACS sorting |
| Database | Primary Data Collection | Organism | Last Updated | URL |
|---|---|---|---|---|
| UTRdb [93] | 5 and 3 UTR regions in EMBL/GenBank records | human, rodent, vertebrate, plant and fungi | 2010 | http://utrdb.ba.itb.cnr.it/ |
| PACdb [95] | cDNA/ESTs | human, mouse, rat, dog, chicken, zebrafish, fugu, fruit fly, mosquito, nematode, Arabidopsis thaliana, rice and baker’s yeast | inaccessible | http://harlequin.jax.org/pacdb/ |
| PolyA_DB [72,94,106] | aligned cDNA/ESTs | human, mouse, rat, chicken and zebrafish | 2018 | http://polya-db.org/v3/ |
| GENCODE Poly (A) site track [100,101] | cDNA/ESTs | human | 2021 | https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=wgEncodeGencodeV19 |
| APADB [97] | MACE-Seq | human, mouse and chicken | 2014 | http://tools.genxpro.net/apadb/ |
| APASdb [96] | SAPAS | human (22 normal and cancer tissues), mouse, zebrafish and some lancelet samples | inaccessible | http://mosas.sysu.edu.cn/utr |
| TC3A [99] | RNA-seq in TCGA | 32 human cancer types | inaccessible | http://tc3a.org/ |
| APAatlas [27] | RNA-seq in GTEx project | >50 human normal tissue | 2020 | https://hanlab.uth.edu/apa/ |
| PolyASite [98] | 3-Seq, 3READS, DRS, QuantSeq_REV, SAPAS, PAPERCLIP, PolyA-seq, PAS-Seq, A-seq, 3P-Seq, DRS, 2P-Seq, PAT-seq | human, mouse and worm | 2020 | https://polyasite.unibas.ch/ |
| Cancer | Gene Markers | Signature APA | Physiological Effects | Molecular Role |
|---|---|---|---|---|
| Breast | PRELID1 | Shortening of 3 UTR | increased protein expression | mitochondrial ROS signalling [139] |
| Breast | SNX3, YME1L1D, USP9X | Shortening of 3 UTR | increased protein levels in short isoform | EGF signalling [22] |
| adult T-cell lymphoma, large B-cell lymphoma, stomach adenocarcinoma | PD-L1 gene (CD274) | Shortening of 3 UTR | PD-1/PD-L1-mediated immune escape in cancer development; structural variants (SVs) disrupt 3 regulatory region of PDL1 | T-cell modulator; PDCD1-mediated inhibitory pathway [136] |
| Colorectal cancer | IGF2BP1/IMP-1 | Shortening of 3 UTR | increased protein levels; increased oncogenic transformation | Modulates pathogenesis [142] |
| TNBC, lung, esophageal, bladder, leukemia, ovarian | N4BP2L2, WDHD1, ZER1, ADGRL2, PRSS12, NPL, SIK3, SYNGR1, SCL2A3, UBE2G2 | Shortening of 3 UTR | unfavourable prognosis | All are related to cancer development: cell cycle regulator and is involved in PI3K/Akt pathway; tumour antigen [138] |
| TNBC | PPIC, ZCCHC14, RTN1, PRCK8, CLIC2, CXCL8, SMAD6 | Lengthening of 3 UTR | poor prognosis; response elements (MREs) in the lengthened 3 UTR leads to homologous gene repression and competing endogenous RNA (ceRNA) resulting in cancer progression; more miRNA binding sites | TGF-βpathway; autocrine NF-ƙB/IL-8 (CXCL8) pathway responsible for cell migration; aberrant pathways and cancer progression [138] |
| TNBC (MB-231) | Caspase 6, DFFA (ICAD), DFFB (CAD), PARP1 | Lengthening of 3 UTR | escape of apoptosis | Caspase pathway [63] |
| TNBC (MB-231) | cyclin D1, D2 | Shortening of 3 UTR | promote cell cycling | Mitotic cell cycle; APC [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandhari, N.; Kraupner-Taylor, C.A.; Harrison, P.F.; Powell, D.R.; Beilharz, T.H. The Detection and Bioinformatic Analysis of Alternative 3′ UTR Isoforms as Potential Cancer Biomarkers. Int. J. Mol. Sci. 2021, 22, 5322. https://doi.org/10.3390/ijms22105322
Kandhari N, Kraupner-Taylor CA, Harrison PF, Powell DR, Beilharz TH. The Detection and Bioinformatic Analysis of Alternative 3′ UTR Isoforms as Potential Cancer Biomarkers. International Journal of Molecular Sciences. 2021; 22(10):5322. https://doi.org/10.3390/ijms22105322
Chicago/Turabian StyleKandhari, Nitika, Calvin A. Kraupner-Taylor, Paul F. Harrison, David R. Powell, and Traude H. Beilharz. 2021. "The Detection and Bioinformatic Analysis of Alternative 3′ UTR Isoforms as Potential Cancer Biomarkers" International Journal of Molecular Sciences 22, no. 10: 5322. https://doi.org/10.3390/ijms22105322
APA StyleKandhari, N., Kraupner-Taylor, C. A., Harrison, P. F., Powell, D. R., & Beilharz, T. H. (2021). The Detection and Bioinformatic Analysis of Alternative 3′ UTR Isoforms as Potential Cancer Biomarkers. International Journal of Molecular Sciences, 22(10), 5322. https://doi.org/10.3390/ijms22105322