
Generation and Characterization of a CRISPR/Cas9-Mediated SNAP29 Knockout in Human Fibroblasts

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
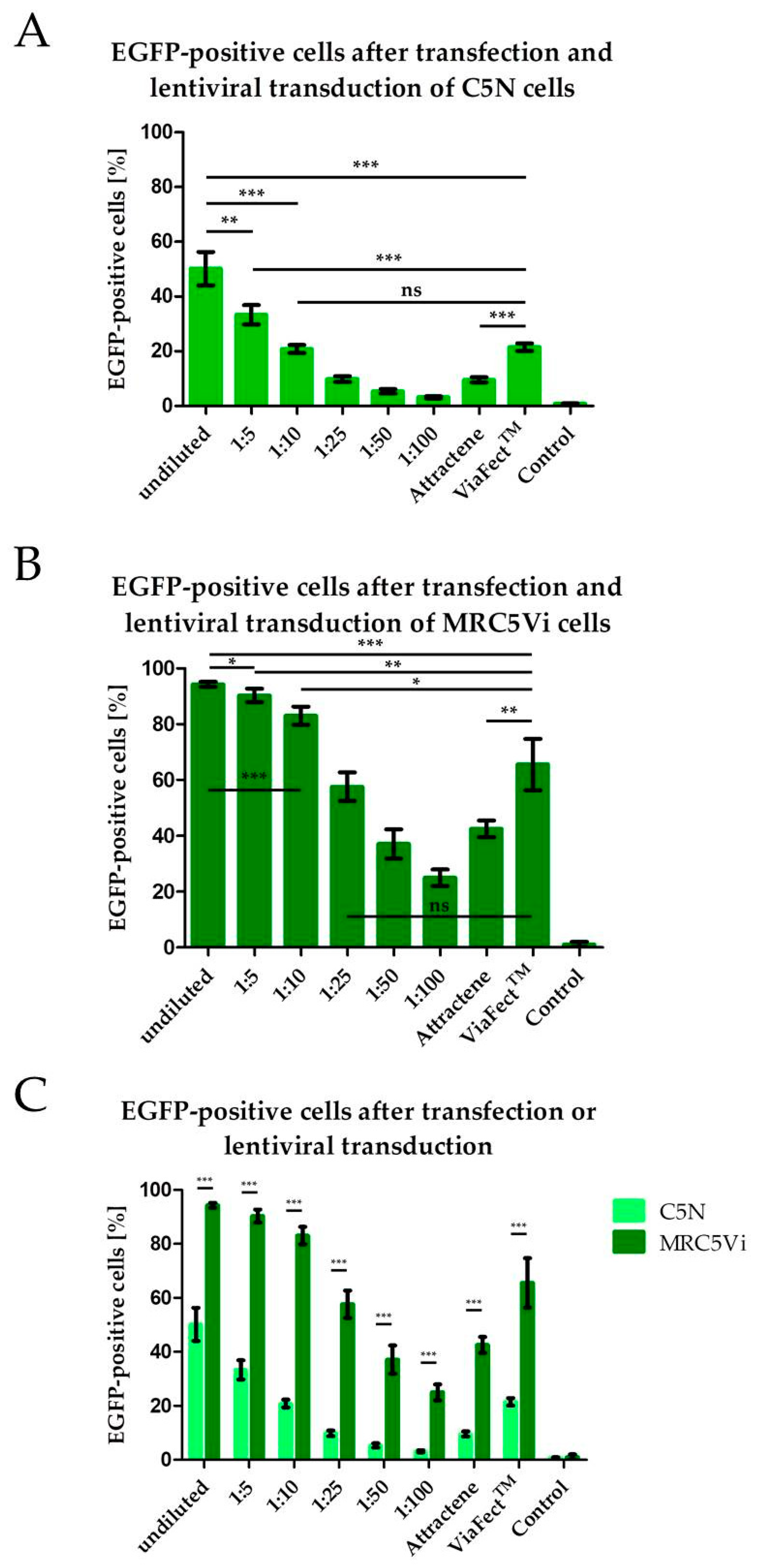
2.1. Efficiency of Lentiviral Transduction and Plasmid Transfection in Different Cell Lines
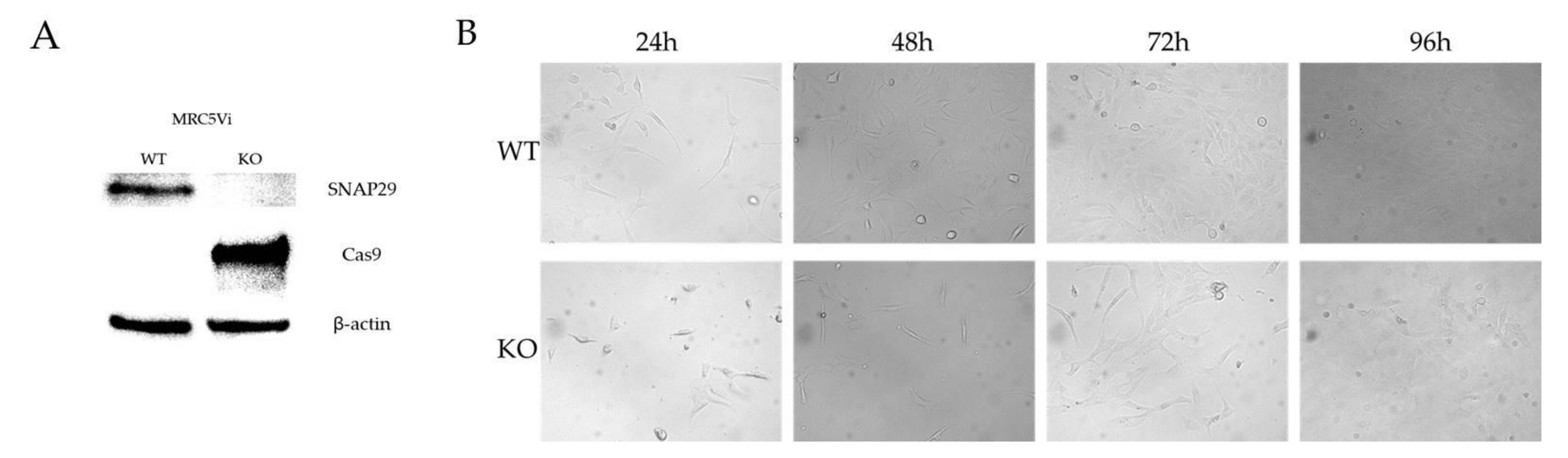
2.2. Generation of a Complete SNAP29 CRISPR/Cas9 KO in MRC5Vi Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentiviral CRISPR Construct Generation
4.3. Lentiviral Vector Production
4.4. Lentiviral Transduction
4.5. Single Clone Expansion
4.6. Sequencing and Clone Evaluation
4.7. Transient Transfection for Flow Cytometry
4.8. Flow Cytometry
4.9. Preparation for Fluorescence Microscopy
4.10. Fluorescence and Bright Field Microscopy
4.11. Western Blot Analysis
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sprecher, E.; Ishida-Yamamoto, A.; Mizrahi-Koren, M.; Rapaport, D.; Goldsher, D.; Indelman, M.; Topaz, O.; Chefetz, I.; Keren, H.; O’Brien, T.J.; et al. A Mutation in SNAP29, Coding for a SNARE Protein Involved in Intracellular Trafficking, Causes a Novel Neurocutaneous Syndrome Characterized by Cerebral Dysgenesis, Neuropathy, Ichthyosis, and Palmoplantar Keratoderma. Am. J. Hum. Genet. 2005, 77, 242–251. [Google Scholar] [CrossRef]
- Hohenstein, A.C.; Roche, P.A. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem. Biophys. Res. Commun. 2001, 285, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Rotem-Yehudar, R.; Galperin, E.; Horowitz, M. Association of Insulin-like Growth Factor 1 Receptor with EHD1 and SNAP29. J. Biol. Chem. 2001, 276, 33054–33060. [Google Scholar] [CrossRef]
- Rapaport, D.; Lugassy, Y.; Sprecher, E.; Horowitz, M. Loss of SNAP29 Impairs Endocytic Recycling and Cell Motility. PLoS ONE 2010, 5, e9759. [Google Scholar] [CrossRef] [PubMed]
- Schiller, S.A.; Seebode, C.; Wieser, G.L.; Goebbels, S.; Ruhwedel, T.; Horowitz, M.; Rapaport, D.; Sarig, O.; Sprecher, E.; Emmert, S. Non-keratinocyte SNAP29 influences epidermal differentiation and hair follicle formation in mice. Exp. Dermatol. 2016, 25, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Schiller, S.A.; Seebode, C.; Wieser, G.L.; Goebbels, S.; Möbius, W.; Horowitz, M.; Sarig, O.; Sprecher, E.; Emmert, S. Establishment of Two Mouse Models for CEDNIK Syndrome Reveals the Pivotal Role of SNAP29 in Epidermal Differentiation. J. Invest. Dermatol. 2016, 136, 672–679. [Google Scholar] [CrossRef]
- Lu, Q.; Insinna, C.; Ott, C.; Stauffer, J.; Pintado, P.A.; Rahajeng, J.; Baxa, U.; Walia, V.; Cuenca, A.; Hwang, Y.-S.; et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat. Cell Biol. 2015, 17, 228–240. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Fuchs-Telem, D.; Stewart, H.; Rapaport, D.; Nousbeck, J.; Gat, A.; Gini, M.; Lugassy, Y.; Emmert, S.; Eckl, K.; Hennies, H.C.; et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br. J. Dermatol. 2011, 164, 610–616. [Google Scholar] [CrossRef]
- Lehmann, J.; Seebode, C.; Emmert, S. Research on genodermatoses using novel genome-editing tools. JDDG J. Ger. Soc. Dermatol. 2017, 15, 783–789. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar]
- Dellambra, E.; Odorisio, T.; D’Arcangelo, D.; Failla, C.M.; Facchiano, A. Non-animal models in dermatological research. ALTEX 2019, 36, 177–202. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 1–13. [Google Scholar] [CrossRef]
- Naldini, L. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr. Opin. Biotechnol. 1998, 9, 457–463. [Google Scholar] [CrossRef]
- Bokhoven, M.; Stephen, S.L.; Knight, S.; Gevers, E.F.; Robinson, I.C.; Takeuchi, Y.; Collins, M.K. Insertional Gene Activation by Lentiviral and Gammaretroviral Vectors. J. Virol. 2009, 83, 283–294. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Kueh, A.J.; Brennan, M.S.; O’Connor, L.; Milla, L.; Wilcox, S.; Tai, L.; Strasser, A.; Herold, M.J. An Inducible Lentiviral Guide RNA Platform Enables the Identification of Tumor-Essential Genes and Tumor-Promoting Mutations In Vivo. Cell Rep. 2015, 10, 1422–1432. [Google Scholar] [CrossRef]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Collins, M.; Radcliffe, P.; Mitrophanous, K.; Takeuchi, Y. Gene transduction efficiency in cells of different species by HIV and EIAV vectors. Gene Ther. 2002, 9, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Addison, C.L.; Hitt, M.; Kunsken, D.; Graham, F.L. Comparison of the human versus murine cytomegalovirus immediate early gene promoters for transgene expression by adenoviral vectors. J. Gen. Virol. 1997, 78, 1653–1661. [Google Scholar] [CrossRef]
- Acevedo, M.L.; García-de Gracia, F.; Miranda-Cárdenas, C.; Soto-Rifo, R.; Aguayo, F.; León, O. Differences in the internalization of self-inactivating VSVG-pseudotyped murine leukemia virus-based vectors in human and murine cells. J. Virol. Methods 2018, 255, 14–22. [Google Scholar] [CrossRef]
- Yamano, S.; Dai, J.; Moursi, A.M. Comparison of Transfection Efficiency of Nonviral Gene Transfer Reagents. Mol. Biotechnol. 2010, 46, 287–300. [Google Scholar] [CrossRef]
- Shi, B.; Xue, M.; Wang, Y.; Wang, Y.; Li, D.; Zhao, X.; Li, X. An improved method for increasing the efficiency of gene transfection and transduction. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 95–104. [Google Scholar]
- Rahmatullah, M.; Jahan, R.; Bashar, A. A review on Berbamine–a Potential Anticancer Drug. World J. Pharm. Pharm. Sci. 2014, 3, 95–110. [Google Scholar]
- Fu, R.; Deng, Q.; Zhang, H.; Hu, X.; Li, Y.; Liu, Y.; Hu, J.; Luo, Q.; Zhang, Y.; Jiang, X.; et al. A novel autophagy inhibitor berbamine blocks SNARE-mediated autophagosome-lysosome fusion through upregulation of BNIP3. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Gerdes, J.M.; Davis, E.E.; Katsanis, N. The Vertebrate Primary Cilium in Development, Homeostasis, and Disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K. V The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Rajput, A.; Martin, I.D.S.; Rose, R.; Beko, A.; LeVea, C.; Sharratt, E.; Mazurchuk, R.; Hoffman, R.M.; Brattain, M.G.; Wang, J. Characterization of HCT116 Human Colon Cancer Cells in an Orthotopic Model. J. Surg. Res. 2008, 147, 276–281. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitive studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Puck, T.T.; Cieciura, S.J.; Robinson, A. Genetics of somatic mammalian cells. III. Long-term cultivation of euploid cells from human and animal subjects. J. Exp. Med. 1958, 108, 945–956. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martens, M.C.; Edelkamp, J.; Seebode, C.; Schäfer, M.; Stählke, S.; Krohn, S.; Jung, O.; Murua Escobar, H.; Emmert, S.; Boeckmann, L. Generation and Characterization of a CRISPR/Cas9-Mediated SNAP29 Knockout in Human Fibroblasts. Int. J. Mol. Sci. 2021, 22, 5293. https://doi.org/10.3390/ijms22105293
Martens MC, Edelkamp J, Seebode C, Schäfer M, Stählke S, Krohn S, Jung O, Murua Escobar H, Emmert S, Boeckmann L. Generation and Characterization of a CRISPR/Cas9-Mediated SNAP29 Knockout in Human Fibroblasts. International Journal of Molecular Sciences. 2021; 22(10):5293. https://doi.org/10.3390/ijms22105293
Chicago/Turabian StyleMartens, Marie Christine, Janin Edelkamp, Christina Seebode, Mirijam Schäfer, Susanne Stählke, Saskia Krohn, Ole Jung, Hugo Murua Escobar, Steffen Emmert, and Lars Boeckmann. 2021. "Generation and Characterization of a CRISPR/Cas9-Mediated SNAP29 Knockout in Human Fibroblasts" International Journal of Molecular Sciences 22, no. 10: 5293. https://doi.org/10.3390/ijms22105293
APA StyleMartens, M. C., Edelkamp, J., Seebode, C., Schäfer, M., Stählke, S., Krohn, S., Jung, O., Murua Escobar, H., Emmert, S., & Boeckmann, L. (2021). Generation and Characterization of a CRISPR/Cas9-Mediated SNAP29 Knockout in Human Fibroblasts. International Journal of Molecular Sciences, 22(10), 5293. https://doi.org/10.3390/ijms22105293