
Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells



Abstract
1. Introduction
2. TNAP Biology
2.1. Cell Biology
2.2. Global Gene Expression and Enzyme Activity
2.3. Gene Expression and Enzyme Activity in BMECs
3. Models and Tools to Study TNAP
3.1. Mouse Models
3.2. TNAP Pharmacological Tools and Pharmaceuticals
4. TNAP in Brain Microvascular Endothelial Health and Function
4.1. TNAP and the Heterogeneity of the Brain’s Vasculature
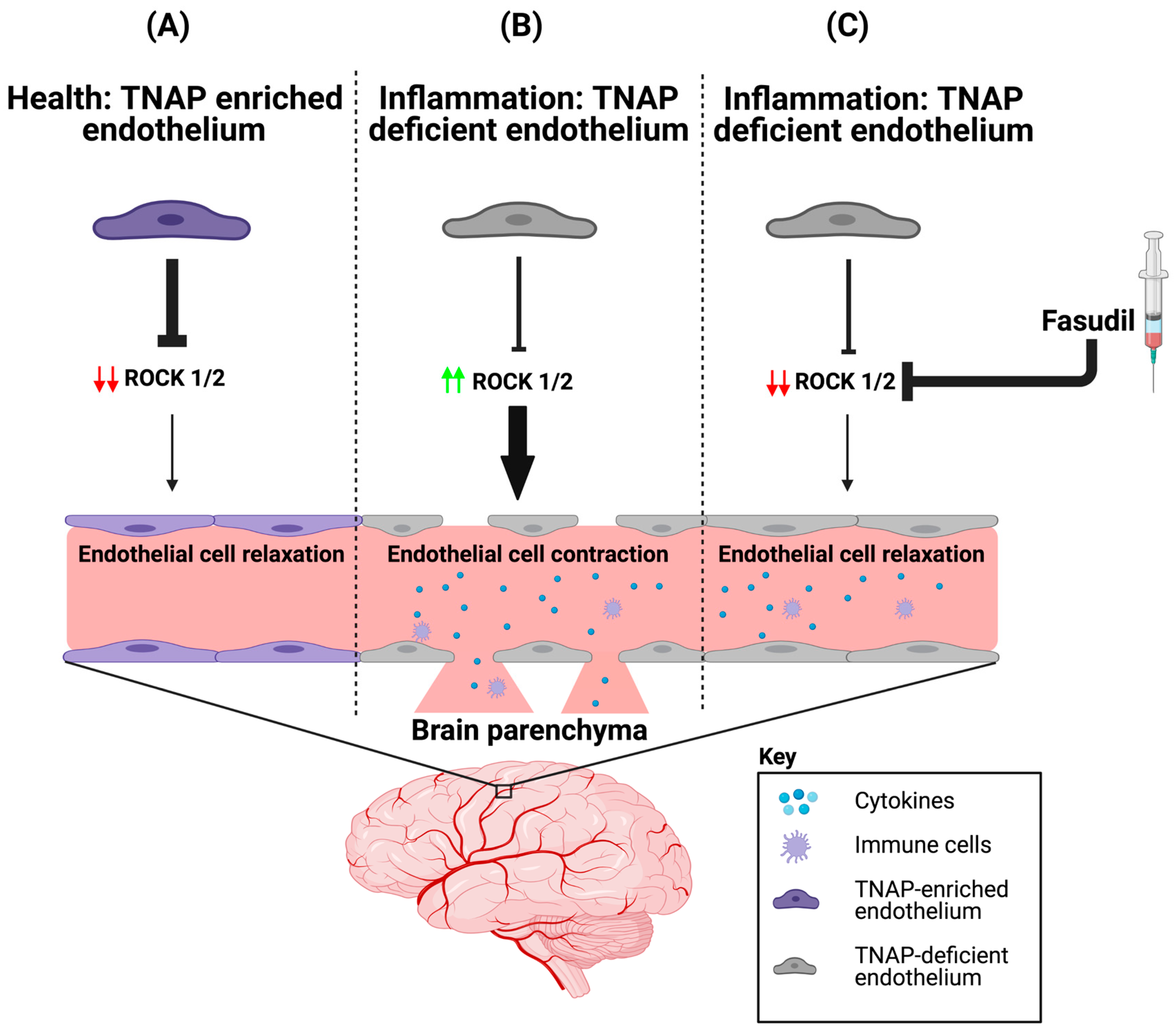
4.2. Potential Mechanism(s) of Action for TNAP in the Brain’s Vasculature
5. A Role for Endothelial TNAP in the Brain’s Microvasculature in Aging and Disease
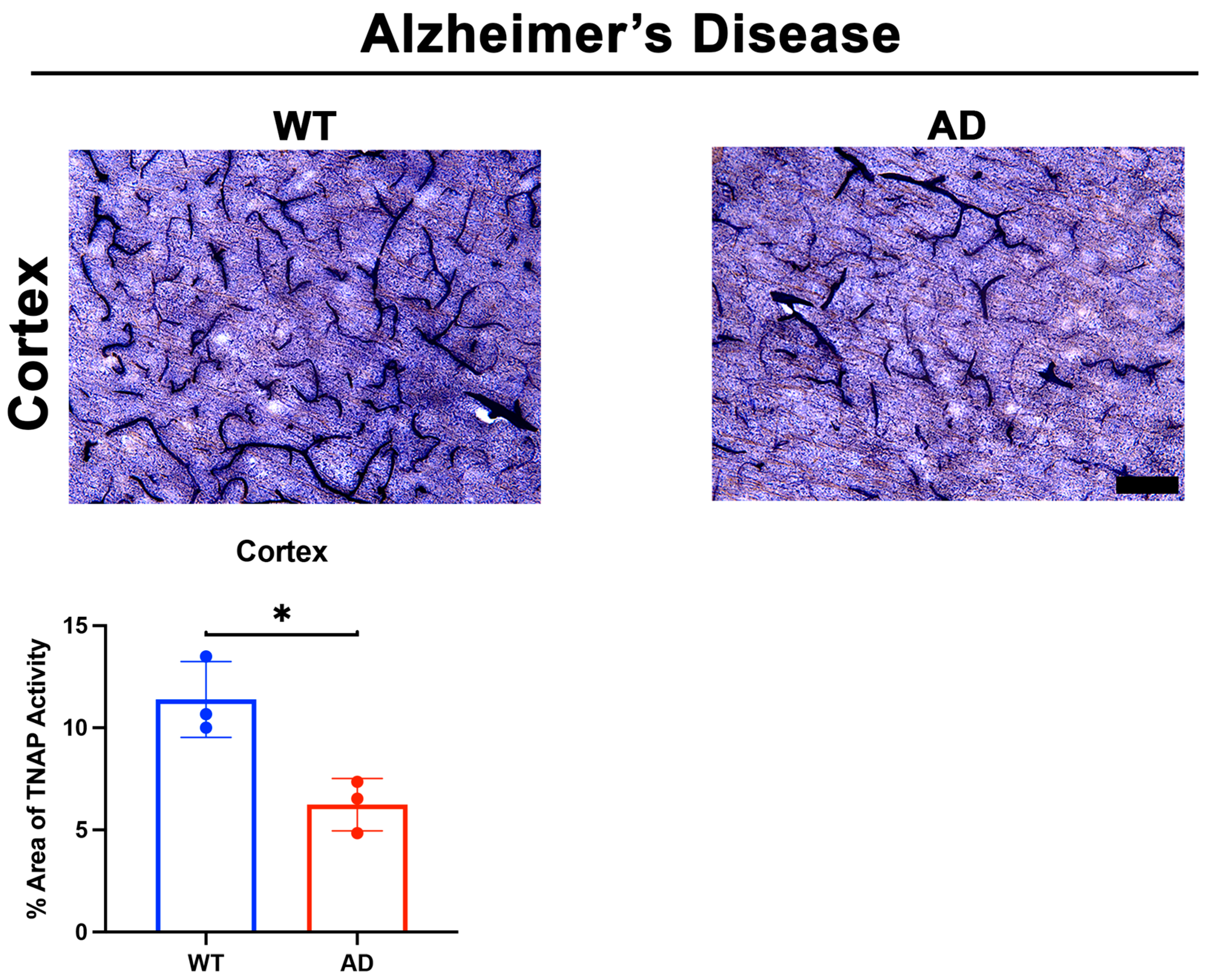
5.1. Brain Microvascular TNAP in Disease
5.2. Brain Microvascular TNAP in Aging
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yu, Q.-J.; Li, M.-C.; Tao, H.; Wang, X. Targeting brain microvascular endothelial cells: A therapeutic approach to neuroprotection against stroke. Neural Regen. Res. 2015, 10, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Liebner, S. Novel insights into the development and maintenance of the blood–brain barrier. Cell Tissue Res. 2014, 355, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Kniesel, U.; Wolburg, H. Tight Junctions of the Blood–Brain Barrier. Cell. Mol. Neurobiol. 2000, 20, 57–76. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Crone, C.; Olesen, S. Electrical resistance of brain microvascular endothelium. Brain Res. 1982, 241, 49–55. [Google Scholar] [CrossRef]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2019, 12, 1019. [Google Scholar] [CrossRef]
- Sedlakova, R.; Shivers, R.R.; Del Maestro, R.F. Ultrastructure of the blood-brain barrier in the rabbit. J. Submicrosc. Cytol. Pathol. 1999, 31, 149–161. [Google Scholar]
- Ayloo, S.; Gu, C. Transcytosis at the blood–brain barrier. Curr. Opin. Neurobiol. 2019, 57, 32–38. [Google Scholar] [CrossRef]
- Nwafor, D.C.; Brichacek, A.L.; Mohammad, A.S.; Griffith, J.; Lucke-Wold, B.P.; Benkovic, S.A.; Geldenhuys, W.J.; Lockman, P.R.; Brown, C.M. Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J. Central Nerv. Syst. Dis. 2019, 11. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial Cells Stimulate Self-Renewal and Expand Neurogenesis of Neural Stem Cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Milner, R. Integrin–Matrix Interactions in the Cerebral Microvasculature. Arter. Thromb. Vasc. Biol. 2006, 26, 1966–1975. [Google Scholar] [CrossRef]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood–brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.; Simonsen, H.; Frederiksen, J.; Rostrup, E.; Larsson, H. Abnormal blood–brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. 2014, 4, 182–189. [Google Scholar] [CrossRef]
- Cramer, S.P.; Simonsen, H.J.; Varatharaj, A.; Galea, I.; Frederiksen, J.L.; Larsson, H.B.W. Permeability of the blood-brain barrier predicts no evidence of disease activity at 2 years after natalizumab or fingolimod treatment in relapsing-remitting multiple sclerosis. Ann. Neurol. 2018, 83, 902–914. [Google Scholar] [CrossRef]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, I.C.M.; De Jong, J.J.A.; Van Boxtel, M.P.J.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Imaging the role of blood–brain barrier disruption in normal cognitive ageing. GeroScience 2020, 42, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T.; Leaver, H.A. Brain Endothelial Cell Death: Modes, Signaling Pathways, and Relevance to Neural Development, Homeostasis, and Disease. Mol. Neurobiol. 2010, 42, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Maudsley, S.; Martin, B. BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004, 27, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.S.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef]
- Breedveld, P.; Beijnen, J.H.; Schellens, J.H. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 2006, 27, 17–24. [Google Scholar] [CrossRef] [PubMed]
- McCandless, E.E.; Piccio, L.; Woerner, B.M.; Schmidt, R.E.; Rubin, J.B.; Cross, A.H.; Klein, R.S. Pathological Expression of CXCL12 at the Blood-Brain Barrier Correlates with Severity of Multiple Sclerosis. Am. J. Pathol. 2008, 172, 799–808. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.S.; Miller, D.S. Tumor Necrosis Factor α and Endothelin-1 Increase P-Glycoprotein Expression and Transport Activity at the Blood-Brain Barrier. Mol. Pharmacol. 2006, 71, 667–675. [Google Scholar] [CrossRef]
- Camire, R.B.; Beaulac, H.J.; Willis, C.L. Transitory loss of glia and the subsequent modulation in inflammatory cytokines/chemokines regulate paracellular claudin-5 expression in endothelial cells. J. Neuroimmunol. 2015, 284, 57–66. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nat. Cell Biol. 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood–brain barrier by targeting endothelial sphingosine 1–phosphate receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef] [PubMed]
- Deracinois, B.; Lenfant, A.-M.; Dehouck, M.-P.; Flahaut, C. Tissue Non-specific Alkaline Phosphatase (TNAP) in Vessels of the Brain. Subcell. Biochem. 2015, 76, 125–151. [Google Scholar] [CrossRef]
- Buchet, R.; Millán, J.L.; Magne, D. Multisystemic Functions of Alkaline Phosphatases. Methods Mol. Biol. 2013, 1053, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Brichacek, A.L.; Benkovic, S.A.; Chakraborty, S.; Nwafor, D.C.; Wang, W.; Jun, S.; Dakhlallah, D.; Geldenhuys, W.J.; Pinkerton, A.B.; Millán, J.L.; et al. Systemic inhibition of tissue-nonspecific alkaline phosphatase alters the brain-immune axis in experimental sepsis. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nwafor, D.C.; Chakraborty, S.; Brichacek, A.L.; Jun, S.; Gambill, C.A.; Wang, W.; Engler-Chiurazzi, E.B.; Dakhlallah, D.; Pinkerton, A.B.; Millán, J.L.; et al. Loss of tissue-nonspecific alkaline phosphatase (TNAP) enzyme activity in cerebral microvessels is coupled to persistent neuroinflammation and behavioral deficits in late sepsis. Brain Behav. Immun. 2020, 84, 115–131. [Google Scholar] [CrossRef]
- Nwafor, D.C.; Brichacek, A.L.; Wang, W.; Bidwai, N.; Lilly, C.L.; Millan, J.; Brown, C.M. Brain endothelial cell tissue-nonspecific alkaline phosphatase (TNAP) activity promotes maintenance of barrier integrity via the ROCK pathway. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nat. Cell Biol. 2020, 583, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G. Biochemistry of the glycosyl-phosphatidylinositol membrane protein anchors. Biochem. J. 1987, 244, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G.; Zilversmit, D.B. Role of phosphatidylinositol in attachment of alkaline phosphatase to membranes. Biochemistry 1980, 19, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.L.; Firth, J.A.; Goldstein, G.W. Polarity of the blood-brain barrier: Distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res. 1980, 192, 17–28. [Google Scholar] [CrossRef]
- Whyte, M.P.; Landt, M.; Ryan, L.M.; Mulivor, R.A.; Henthorn, P.S.; Fedde, K.N.; Mahuren, J.D.; Coburn, S.P. Alkaline phosphatase: Placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5’-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J. Clin. Investig. 1995, 95, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Say, J.; Ciuffi, K.; Furriel, R.P.; Ciancaglini, P.; Leone, F.A. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta BBA Gen. Subj. 1991, 1074, 256–262. [Google Scholar] [CrossRef]
- Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.A.; Ward, J.C.; Rivas, M.L.; Magill, H.L.; Whyte, M.P. Infantile hypophosphatasia: Autosomal recessive transmission to two related sibships. Am. J. Med Genet. 1990, 36, 15–22. [Google Scholar] [CrossRef]
- Barvencik, F.; Beil, F.T.; Gebauer, M.; Busse, B.; Koehne, T.; Seitz, S.; Zustin, J.; Pogoda, P.; Schinke, T.; Amling, M. Skeletal mineralization defects in adult hypophosphatasia—a clinical and histological analysis. Osteoporos. Int. 2011, 22, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millan, J.L.; MacGregor, G.R.; et al. Alkaline Phosphatase Knock-Out Mice Recapitulate the Metabolic and Skeletal Defects of Infantile Hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia and the Role of Alkaline Phosphatase in Skeletal Mineralization. Endocr. Rev. 1994, 15, 439–461. [Google Scholar] [CrossRef]
- Orimo, H. The Mechanism of Mineralization and the Role of Alkaline Phosphatase in Health and Disease. J. Nippon. Med. Sch. 2010, 77, 4–12. [Google Scholar] [CrossRef]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010, 1192, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef]
- Whyte, M.P.; Mahuren, J.D.; Vrabel, L.A.; Coburn, S.P. Markedly increased circulating pyridoxal-5’-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J. Clin. Investig. 1985, 76, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Brun-Heath, I.; Ermonval, M.; Chabrol, E.; Xiao, J.; Palkovits, M.; Lyck, R.; Miller, F.; Couraud, P.-O.; Mornet, E.; Fonta, C. Differential expression of the bone and the liver tissue non-specific alkaline phosphatase isoforms in brain tissues. Cell Tissue Res. 2010, 343, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Toh, Y.; Yamamoto, M.; Endo, H.; Misumi, Y.; Ikehara, Y. Isolation and characterization of a rat liver alkaline phosphatase gene. A single gene with two promoters. JBIC J. Biol. Inorg. Chem. 1989, 182, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Terao, M.; Gianni, M.; Garattini, E. Characterization of a second promoter for the mouse liver/bone/kidney-type alkaline phosphatase gene: Cell and tissue specific expression. Biochem. Biophys. Res. Commun. 1991, 179, 1352–1360. [Google Scholar] [CrossRef]
- Matsuura, S.; Kishi, F.; Kajii, T. Characterization of a 5′-flanking region of the human liver/bone/kidney alkaline phosphatase gene: Two kinds of mRNA from a single gene. Biochem. Biophys. Res. Commun. 1990, 168, 993–1000. [Google Scholar] [CrossRef]
- Hahnel, A.; Rappolee, D.; Millan, J.; Manes, T.; Ziomek, C.; Theodosiou, N.; Werb, Z.; Pedersen, R.; Schultz, G. Two alkaline phosphatase genes are expressed during early development in the mouse embryo. Development 1990, 110, 555–564. [Google Scholar] [CrossRef]
- Kruse, K.; Hanefeld, F.; Kohlschütter, A.; Rosskamp, R.; Gross-Selbeck, G. Hyperphosphatasia with mental retardation. J. Pediatr. 1988, 112, 436–439. [Google Scholar] [CrossRef]
- Thompson, M.D.; Nezarati, M.M.; Gillessen-Kaesbach, G.; Meinecke, P.; Mendoza, R.; Mornet, E.; Brun-Heath, I.; Squarcioni, C.P.; Legeai-Mallet, L.; Munnich, A.; et al. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am. J. Med. Genet. Part A 2010, 152A, 1661–1669. [Google Scholar] [CrossRef]
- Thompson, M.D.; Cole, D.E.; Mabry, C.C. 50 Years Ago in T J P. J. Pediatr. 2020, 222, 97. [Google Scholar] [CrossRef]
- Anstrom, J.A.; Brown, W.R.; Moody, D.M.; Thore, C.R.; Challa, V.R.; Block, S.M. Temporal expression pattern of cerebrovascular endothelial cell alkaline phosphatase during human gestation. J. Neuropathol. Exp. Neurol. 2002, 61, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N. Histochemical studies on the phosphatase of the nervous system. J. Comp. Neurol. 1950, 93, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Fonta, C.; Imbert, M. Vascularization in the Primate Visual Cortex during Development. Cereb. Cortex 2002, 12, 199–211. [Google Scholar] [CrossRef]
- Bell, M.A.; Scarrow, W.G. Staining for microvascular alkaline phosphatase in thick celloidin sections of nervous tissue: Morphometric and pathological applications. Microvasc. Res. 1984, 27, 189–203. [Google Scholar] [CrossRef]
- Norman, M.G.; O’Kusky, J.R. The growth and development of microvasculature in human cerebral cortex. J. Neuropathol. Exp. Neurol. 1986, 45, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Latker, C.H.; Shinowara, N.L.; Miller, J.C.; Rapoport, S.I. Differential localization of alkaline phosphatase in barrier tissues of the frog and rat nervous systems: A cytochemical and biochemical study. J. Comp. Neurol. 1987, 264, 291–302. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Hashioka, Y.; Utoguchi, N.; Kubo, K.; Nakagawa, S.; Mayumi, T. A Comparison of Drug Transport through Cultured Monolayers of Bovine Brain Capillary and Bovine Aortic Endothelial Cells. Biol. Pharm. Bull. 1994, 17, 1385–1390. [Google Scholar] [CrossRef]
- Vorbrodt, A.W.; Lossinsky, A.S.; Wisniewski, H.M. Localization of Alkaline Phosphatase Activity in Endothelia of Developing and Mature Mouse Blood-Brain Barrier. Dev. Neurosci. 1986, 8, 1–13. [Google Scholar] [CrossRef]
- Mayahara, H.; Hirano, H.; Saito, T.; Ogawa, K. The new lead citrate method for the ultracytochemical demonstration of activity of non-specific alkaline phosphatase (orthophosphoric monoester phosphohydrolase). Histochem. Cell Biol. 1967, 11, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Nagano, M. Electron-microscopic cytochemistry of alkaline-phosphatase activity in endothelium, pericytes and oligodendrocytes in the rat brain. Histochem. Cell Biol. 1985, 82, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ovtscharoff, W. Ultracytochemische Lokalisierung der alkalischen Phosphatase im Cortex cerebri bei neugeborenen Ratten. Histochem. Cell Biol. 1973, 37, 93–95. [Google Scholar] [CrossRef]
- Deracinois, B.; Duban-Deweer, S.; Pottiez, G.; Cecchelli, R.; Karamanos, Y.; Flahaut, C. TNAP and EHD1 Are Over-Expressed in Bovine Brain Capillary Endothelial Cells after the Re-Induction of Blood-Brain Barrier Properties. PLoS ONE 2012, 7, e48428. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Rauh, J.; Galla, H.-J. The Susceptibility of Cerebral Endothelial Cells to Astroglial Induction of Blood-Brain Barrier Enzymes Depends on Their Proliferative State. J. Neurochem. 1991, 57, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Rauh, J.; Meyer, J.; Beuckmann, C.; Galla, H.-J. Chapter 18: Development of an in vitro cell culture system to mimic the blood-brain barrier. Progress Brain Res. 1992, 91, 117–121. [Google Scholar] [CrossRef]
- Tio, S.; Deenen, M.; Marani, E. Astrocyte-mediated induction of alkaline phosphatase activity in human umbilical cord vein endothelium: An in vitro model. Eur. J. Morphol. 1990, 28, 289–300. [Google Scholar] [PubMed]
- Ohlebusch, B.; Borst, A.; Frankenbach, T.; Klopocki, E.; Jakob, F.; Liedtke, D.; Graser, S. Investigation of alpl expression and Tnap-activity in zebrafish implies conserved functions during skeletal and neuronal development. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- McComb, R.B.; Bowers, G.N., Jr.; Posen, S. Alkaline Phosphatase; Springer Science and Business Media LLC: Berlin, Germany, 1979; 986p. [Google Scholar]
- Friede, R.L. A quantitative mapping of alkaline phosphatase in the brain of the rhesus monkey. J. Neurochem. 1966, 13, 197–203. [Google Scholar] [CrossRef]
- Negyessy, L.; Xiao, J.; Kántor, O.; Kovacs, G.G.; Palkovits, M.; Doczi, T.; Renaud, L.; Baksa, G.; Glasz, T.; Ashaber, M.; et al. Layer-specific activity of tissue non-specific alkaline phosphatase in the human neocortex. Neuroscience 2011, 172, 406–418. [Google Scholar] [CrossRef]
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.D.S.; Millán, J.L.; Hatch, N.E. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl−/− mouse model of infantile hypophosphatasia. Bone 2014, 67, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, D.; Hofmann, C.; Jakob, F.; Klopocki, E.; Graser, S. Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules 2020, 10, 1648. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Serrano, Á.; De Diego-García, L.; Henshall, D.C.; Engel, T.; Diaz-Hernandez, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Belmonte, R.; Tena-Garitaonaindia, M.; Hernández-Chirlaque, C.; Córdova, S.; Ceacero-Heras, D.; De Medina, F.S.; Martínez-Augustin, O. Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. Int. J. Mol. Sci. 2020, 22, 51. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Gámez-Belmonte, R.; Ocón, B.; Martínez-Moya, P.; Wirtz, S.; de Medina, F.S.; Martínez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohns Colitis 2016, 11, 857–870. [Google Scholar] [CrossRef][Green Version]
- Yamamoto, S.; Orimo, H.; Matsumoto, T.; Iijima, O.; Narisawa, S.; Maeda, T.; Millán, J.L.; Shimada, T. Prolonged survival and phenotypic correction of Akp2−/− hypophosphatasia mice by lentiviral gene therapy. J. Bone Miner. Res. 2010, 26, 135–142. [Google Scholar] [CrossRef]
- Nam, H.K.; Vesela, I.; Schutte, S.D.; Hatch, N.E. Viral delivery of tissue nonspecific alkaline phosphatase diminishes craniosynostosis in one of two FGFR2C342Y/+ mouse models of Crouzon syndrome. PLoS ONE 2020, 15, e0234073. [Google Scholar] [CrossRef]
- Arnò, B.; Galli, F.; Roostalu, U.; Aldeiri, B.M.; Miyake, T.; Albertini, A.; Bragg, L.; Prehar, S.; McDermott, J.C.; Cartwright, E.J.; et al. TNAP limits TGF-β-dependent cardiac and skeletal muscle fibrosis by inactivating the SMAD2/3 transcription factors. J. Cell Sci. 2019, 132, jcs.234948. [Google Scholar] [CrossRef]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.A.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Hear. Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef]
- Rodionov, R.N.; Begmatov, H.; Jarzebska, N.; Patel, K.; Mills, M.T.; Ghani, Z.; Khakshour, D.; Tamboli, P.; Patel, M.N.; Abdalla, M.; et al. Homoarginine Supplementation Prevents Left Ventricular Dilatation and Preserves Systolic Function in a Model of Coronary Artery Disease. J. Am. Hear. Assoc. 2019, 8, e012486. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, F.; Corbo, A.; Salehi, M.; Yadav, M.C.; Salman, S.; Petrosian, D.; Rashidbaigi, O.J.; Chait, J.; Kuruvilla, J.; Plummer, M.; et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS ONE 2017, 12, e0186426. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.; Kuss, P.; Yadav, M.; Kolli, T.; Narisawa, S.; Lukashova, L.; Cory, E.; Sah, R.; Somerman, M.; Millán, J. Conditional Alpl Ablation Phenocopies Dental Defects of Hypophosphatasia. J. Dent. Res. 2016, 96, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Alva, J.A.; Zovein, A.C.; Monvoisin, A.; Murphy, T.; Salazar, A.; Harvey, N.L.; Carmeliet, P.; Iruela-Arispe, M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006, 235, 759–767. [Google Scholar] [CrossRef]
- Assmann, J.C.; Körbelin, J.; Schwaninger, M. Genetic manipulation of brain endothelial cells in vivo. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 381–394. [Google Scholar] [CrossRef]
- Rufo, M.B.; Fishman, W.H. l-Homoarginine, a specific inhibitor of liver-type alkaline phosphatase, applied to the recognition of liver-type enzyme activity in rat intestine. J. Histochem. Cytochem. 1972, 20, 336–343. [Google Scholar] [CrossRef]
- Kozlenkov, A.; Le Du, M.H.; Cuniasse, P.; Ny, T.; Hoylaerts, M.F.; Millán, J.L. Residues Determining the Binding Specificity of Uncompetitive Inhibitors to Tissue-Nonspecific Alkaline Phosphatase. J. Bone Miner. Res. 2004, 19, 1862–1872. [Google Scholar] [CrossRef]
- Dahl, R.; Sergienko, E.A.; Su, Y.; Mostofi, Y.S.; Yang, L.; Simão, A.M.; Narisawa, S.; Brown, B.; Mangravita-Novo, A.; Vicchiarelli, M.; et al. Discovery and Validation of a Series of Aryl Sulfonamides as Selective Inhibitors of Tissue-Nonspecific Alkaline Phosphatase (TNAP). J. Med. Chem. 2009, 52, 6919–6925. [Google Scholar] [CrossRef]
- Nakamura, T.; Nakamura-Takahashi, A.; Kasahara, M.; Yamaguchi, A.; Azuma, T. Tissue-nonspecific alkaline phosphatase promotes the osteogenic differentiation of osteoprogenitor cells. Biochem. Biophys. Res. Commun. 2020, 524, 702–709. [Google Scholar] [CrossRef]
- Pinkerton, A.B.; Sergienko, E.; Bravo, Y.; Dahl, R.; Ma, C.-T.; Sun, Q.; Jackson, M.R.; Cosford, N.D.P.; Millán, J.L. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 31–34. [Google Scholar] [CrossRef]
- Genest, F.; Rak, D.; Petryk, A.; Seefried, L. Physical Function and Health-Related Quality of Life in Adults Treated With Asfotase Alfa for Pediatric-Onset Hypophosphatasia. JBMR Plus 2020, 4. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Rockman-Greenberg, C.; Rauch, F.; Bhatti, M.T.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P. Five-year efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone 2019, 121, 149–162. [Google Scholar] [CrossRef]
- Whyte, M.P.; Simmons, J.H.; Moseley, S.; Fujita, K.P.; Bishop, N.; Salman, N.J.; Taylor, J.; Phillips, D.; McGinn, M.; McAlister, W.H. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes Endocrinol. 2019, 7, 93–105. [Google Scholar] [CrossRef]
- Pickkers, P.; Snellen, F.; Rogiers, P.; Bakker, J.; Jorens, P.; Meulenbelt, J.; Spapen, H.; Tulleken, J.E.; Lins, R.; Ramael, S.; et al. Clinical pharmacology of exogenously administered alkaline phosphatase. Eur. J. Clin. Pharmacol. 2009, 65, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, S.; Masereeuw, R.; Moesker, O.; Bouw, M.P.W.J.M.; Van Der Hoeven, J.G.; Peters, W.H.M.; Russel, F.G.M.; Pickkers, P. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit. Care Med. 2009, 37, 417-e1. [Google Scholar] [CrossRef] [PubMed]
- Bender, B.; Baranyi, M.; Kerekes, A.; Bodrogi, L.; Brands, R.; Uhrin, P.; Bösze, Z. Recombinant Human Tissue Non-Specific Alkaline Phosphatase Successfully Counteracts Lipopolysaccharide Induced Sepsis in Mice. Physiol. Res. 2015, 64, 731–738. [Google Scholar] [CrossRef]
- Pickkers, P.; Heemskerk, S.; Schouten, J.; Laterre, P.-F.; Vincent, J.-L.; Beishuizen, A.; Jorens, P.G.; Spapen, H.; Bulitta, M.; Peters, W.H.; et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: A prospective randomized double-blind placebo-controlled trial. Crit. Care 2012, 16, R14. [Google Scholar] [CrossRef] [PubMed]
- Lukas, M.; Drastich, P.; Konecny, M.; Gionchetti, P.; Urban, O.; Cantoni, F.; Bortlik, M.; Duricova, D.; Bulitta, M. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm. Bowel Dis. 2010, 16, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Adebayo, D.; Oria, M.; De Chiara, F.; Novelli, S.; Habtesion, A.; Davies, N.; Andreola, F.; Jalan, R. Recombinant Alkaline Phosphatase Prevents Acute on Chronic Liver Failure. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Peters, E.; Ergin, B.; Kandil, A.; Gurel-Gurevin, E.; Van Elsas, A.; Masereeuw, R.; Pickkers, P.; Ince, C. Effects of a human recombinant alkaline phosphatase on renal hemodynamics, oxygenation and inflammation in two models of acute kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Juschten, J.; for the BASIC study investigators; Ingelse, S.A.; Bos, L.D.J.; Girbes, A.R.J.; Juffermans, N.P.; Van Der Poll, T.; Schultz, M.J.; Tuinman, P.R. Alkaline phosphatase in pulmonary inflammation—A translational study in ventilated critically ill patients and rats. Intensive Care Med. Exp. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Pickkers, P.; Mehta, R.L.; Murray, P.T.; Joannidis, M.; Molitoris, B.A.; Kellum, J.A.; Bachler, M.; Hoste, E.A.J.; Hoiting, O.; Krell, K.; et al. Effect of Human Recombinant Alkaline Phosphatase on 7-Day Creatinine Clearance in Patients With Sepsis-Associated Acute Kidney Injury. JAMA 2018, 320, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nat. Cell Biol. 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Beuckmann, C.; Hellwig, S.; Galla, H.-J. Induction of the Blood/Brain-Barrier-Associated Enzyme Alkaline Phosphatase in Endothelial Cells from Cerebral Capillaries is Mediated Via cAMP. JBIC J. Biol. Inorg. Chem. 1995, 229, 641–644. [Google Scholar] [CrossRef]
- Roux, F.; Durieu-Trautmann, O.; Chaverot, N.; Claire, M.; Mailly, P.; Bourre, J.-M.; Strosberg, A.D.; Couraud, P.-O. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J. Cell. Physiol. 1994, 159, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.-I.; Ohtomo, E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, T.; Asai, T.; Yanagida, Y.; Namba, M.; Koide, H.; Shimizu, K.; Oku, N. Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke. FASEB J. 2017, 31, 1879–1890. [Google Scholar] [CrossRef]
- Liu, K.; Li, Z.; Wu, T.; Ding, S. Role of Rho Kinase in Microvascular Damage Following Cerebral Ischemia Reperfusion in Rats. Int. J. Mol. Sci. 2011, 12, 1222–1231. [Google Scholar] [CrossRef]
- Jianjun, Z.; Baochun, Z.; Limei, M.; Lijun, L. Exploring the beneficial role of ROCK inhibitors in sepsis-induced cerebral and cognitive injury in rats. Fundam. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Adams, S.E.; Melnykovych, G. Synergistic stimulation of alkaline phosphatase activity in bovine aortic endothelial cells grown in the presence of retinoids and glucocorticoids. J. Cell. Physiol. 1985, 124, 120–124. [Google Scholar] [CrossRef]
- Nakazato, H.; Deguchi, M.; Fujimoto, M.; Fukushima, H. Alkaline phosphatase expression in cultured endothelial cells of aorta and brain micro vessels: Induction by interleukin-6-type cytokines and suppression by transforming growth factor betas. Life Sci. 1997, 61, 2065–2072. [Google Scholar] [CrossRef]
- Romero, C.R.; Herzig, D.S.; Etogo, A.; Nunez, J.; Mahmoudizad, R.; Fang, G.; Murphey, E.D.; Toliver-Kinsky, T.; Sherwood, E.R. The role of interferon-γ in the pathogenesis of acute intra-abdominal sepsis. J. Leukoc. Biol. 2010, 88, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Moody, D.M.; Thore, C.R.; Challa, V.R.; Anstrom, J.A. Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J. Neurol. Sci. 2007, 257, 62–66. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 1–27. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term Cognitive Impairment and Functional Disability Among Survivors of Severe Sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef]
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; Huerta, P.T.; Robbiati, S.; Valdes-Ferrer, S.I.; Ochani, M.; Dancho, M.; Frankfurt, M.; Volpe, B.T.; Tracey, K.J.; Diamond, B. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol. Med. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.R.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor Necrosis Factor-α. Stroke 1997, 28, 1233–1244. [Google Scholar] [CrossRef]
- Michie, H.R.; Manogue, K.R.; Spriggs, D.R.; Revhaug, A.; O’Dwyer, S.; Dinarello, C.A.; Cerami, A.; Wolff, S.M.; Wilmore, D.W. Detection of Circulating Tumor Necrosis Factor after Endotoxin Administration. N. Engl. J. Med. 1988, 318, 1481–1486. [Google Scholar] [CrossRef]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T Lymphocytes and Interferon-γ in Ischemic Stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef]
- Colton, C.A.; Wilcock, D.M.; Wink, D.A.; Davis, J.; Van Nostrand, W.E.; Vitek, M.P. The Effects of NOS2 Gene Deletion on Mice Expressing Mutated Human AβPP. J. Alzheimer’s Dis. 2008, 15, 571–587. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Lewis, M.R.; Van Nostrand, W.E.; Davis, J.; Previti, M.L.; Gharkholonarehe, N.; Vitek, M.P.; Colton, C.A. Progression of Amyloid Pathology to Alzheimer’s Disease Pathology in an Amyloid Precursor Protein Transgenic Mouse Model by Removal of Nitric Oxide Synthase 2. J. Neurosci. 2008, 28, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Nwafor, D.C.; Chakraborty, S.; Jun, S.; Brichacek, A.L.; Dransfeld, M.; Gemoets, D.E.; Dakhlallah, D.; Brown, C.M. Disruption of metabolic, sleep, and sensorimotor functional outcomes in a female transgenic mouse model of Alzheimer’s disease. Behav. Brain Res. 2021, 398, 112983. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, M.; Gómez-Ramos, A.; Rubio, A.; Gómez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific Alkaline Phosphatase Promotes the Neurotoxicity Effect of Extracellular Tau. J. Biol. Chem. 2010, 285, 32539–32548. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood–brain barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000. [Google Scholar] [CrossRef]
- Chen, M.B.; Yang, A.C.; Yousef, H.; Lee, D.; Chen, W.; Schaum, N.; Lehallier, B.; Quake, S.R.; Wyss-Coray, T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020, 30, 4418–4432. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Animal Models | TNAP Activity in Cerebral Microvessels | Gene Name | Time Period When First Detected in CNS | Ref. |
|---|---|---|---|---|
| Zebrafish | Activity is present in the brain parenchyma and vessels | alpl | 11 h post-fertiilzation (hpf) | [78] |
| Mouse | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl or Akp2 | Embryonic day 7–14 | [59,64] |
| Rat | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl or Akp2 | Embryonic day 15 | [54,64] |
| Guinea Pig | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl | - | [64] |
| Frog | Activity is absent on vessels but present on inner arachnoid and perineurium | alpl | - | [68] |
| Chicken | Weak activity in vessels and strong activity in the brain parenchyma | ALPL | Day 2 of the incubation period | [64,79] |
| Rabbit | Activity is absent in vessels but strong in brain parenchyma | ALPL | - | [64] |
| Cat | Strong activity in vessels and weaker activity in the brain parenchyma | ALPL | - | [64] |
| Rhesus Monkey | Strong activity in vessels and laminar activity pattern in the brain parenchyma (i.e., some areas/layers have higher activity than others) | ALPL | - | [80] |
| Human | Strong activity in vessels and laminar activity pattern in the brain parenchyma (i.e., some areas/layers have higher activity than others) | ALPL | 28 weeks of gestation | [63,81] |
| Mouse Model Type | Cre-Mediated Target Cell | Preclinical Outcomes | Ref. |
|---|---|---|---|
| TNAP OE | Vascular smooth muscle cells (Tagln-Cre or SM22-Cre) | Mice had increased AP enzyme activity, increased systolic blood pressure, and increased vascular calcification. Male TNAP-OE mice lifespan was shorter than WT controls, as all died by 5 months of age. | [90] |
| TNAP OE | Endothelial cells (Tie2-Cre) | Increased AP activity was localized to the luminal side of the aorta and vascular networks of heart, lung, kidney, liver, small intestine, and pancreas. No skeletal abnormalities were detected; however, in the heart, kidney, mesentery, pancreas, spleen, and lung parenchyma there were calcified lesions in adult mice. | [91] |
| TNAP OE with “wicked high cholesterol” (C57BL/6J-LdlrHlb301/J) | Endothelial cells (Tie2-Cre) | Mice displayed increased AP activity in endothelial cells and increased sub-endothelial calcification nodules in their coronary arteries, which recapitulates murine atherosclerosis. | [92,93] |
| TNAP OE | Endothelial cells (Cdh5-Cre) | Over-expression of TNAP on endothelial cells resulted in increased survival and decreased clinical severity post-sepsis compared to controls. Locomotor activity in the last 5 min of open field testing was also increased in VE-cOE mice compared to controls. | [35] |
| TNAP KO | Osteoblasts and odontoblasts (Col1a1-Cre); Early limb bud mesenchyme and a subset of craniofacial mesenchyme (Prx1-Cre) | While both Cre recombinase drivers result in similar phenotypes with regards to skeletal defects in cortical and trabecular bone, Prx1-cKO mouse long bones appeared more severely affected. Both models resulted in increased osteoclast numbers on alveolar bone surfaces and reduced alveolar bone height. Both models resulted in cementum and periodontal ligament defects, consistent with periodontal disease. | [94] |
| TNAP KO | Endothelial cells (Cdh5-Cre) | Primary BMECs revealed decreased barrier integrity compared to controls, which was mitigated after treatment with fasudil. | [37] (results reported in a preprint) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwafor, D.C.; Brichacek, A.L.; Ali, A.; Brown, C.M. Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 5257. https://doi.org/10.3390/ijms22105257
Nwafor DC, Brichacek AL, Ali A, Brown CM. Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(10):5257. https://doi.org/10.3390/ijms22105257
Chicago/Turabian StyleNwafor, Divine C., Allison L. Brichacek, Ahsan Ali, and Candice M. Brown. 2021. "Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells" International Journal of Molecular Sciences 22, no. 10: 5257. https://doi.org/10.3390/ijms22105257
APA StyleNwafor, D. C., Brichacek, A. L., Ali, A., & Brown, C. M. (2021). Tissue-Nonspecific Alkaline Phosphatase in Central Nervous System Health and Disease: A Focus on Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences, 22(10), 5257. https://doi.org/10.3390/ijms22105257