
High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures


 ,
,  ,
,  and
and 
Abstract

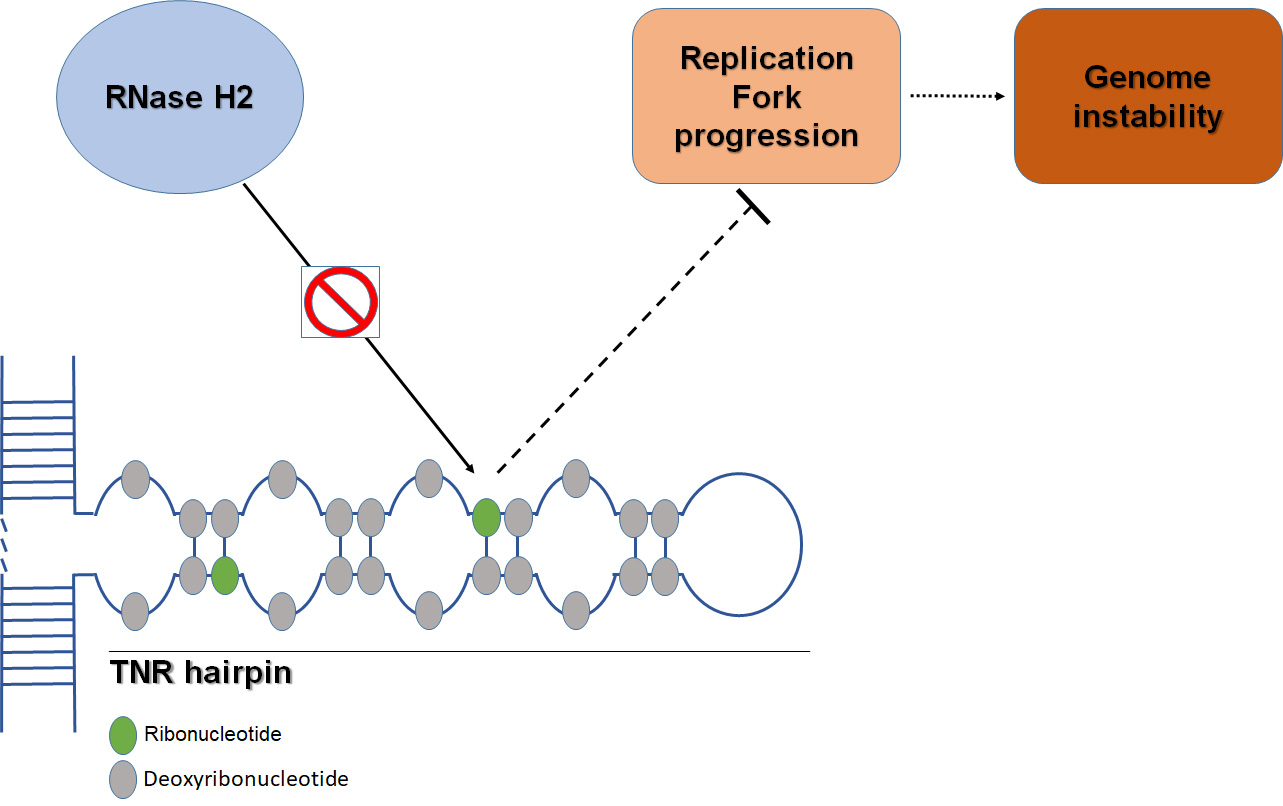
1. Introduction
2. Results
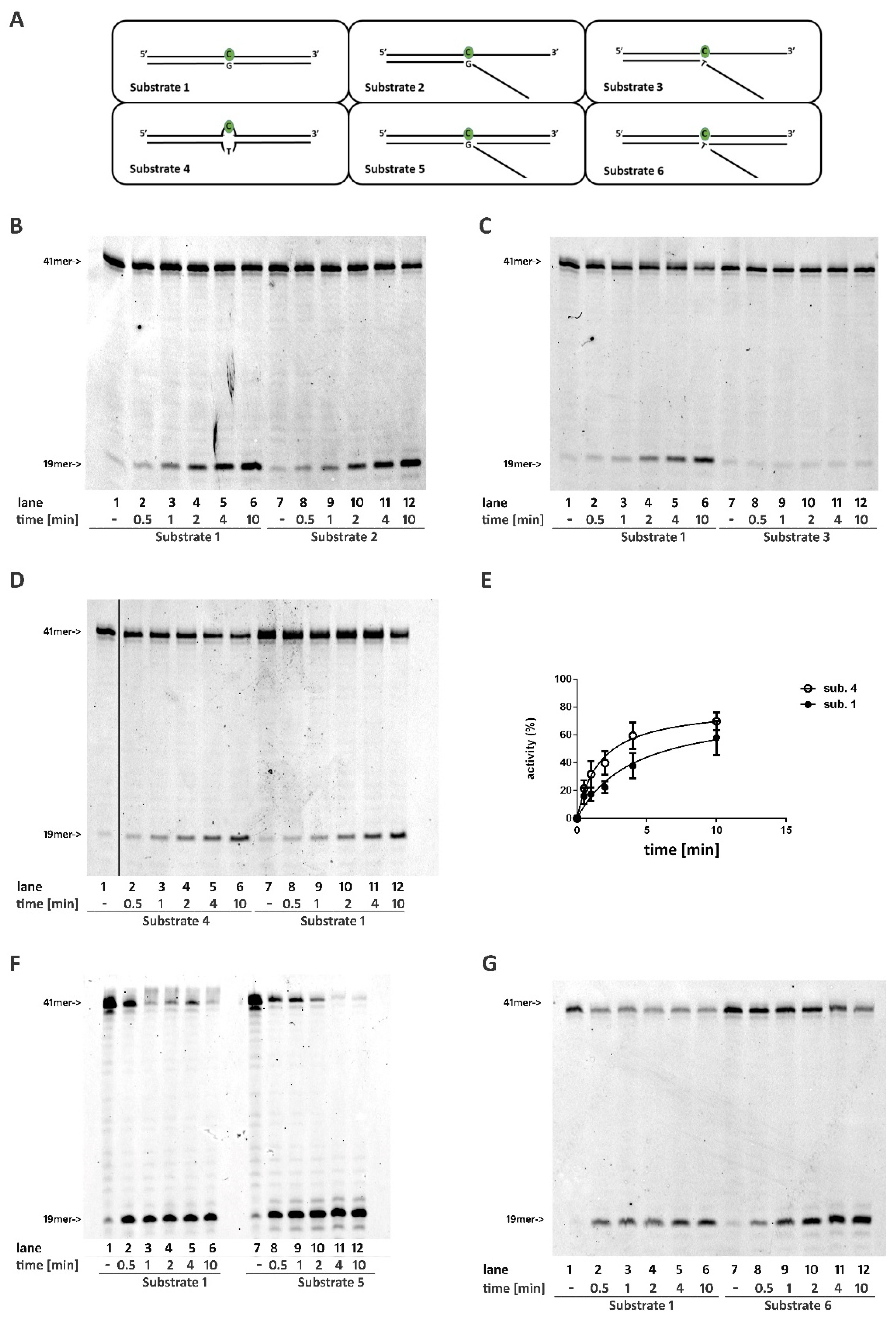
2.1. A DNA Double Stranded Structure at the 3′ of the Ribonucleotide Is Not Required for RNase H2 Activity
2.2. RNase H2 Can Process a Mismatched Ribonucleotide Embedded within an Intact DNA Double-Stranded Structure
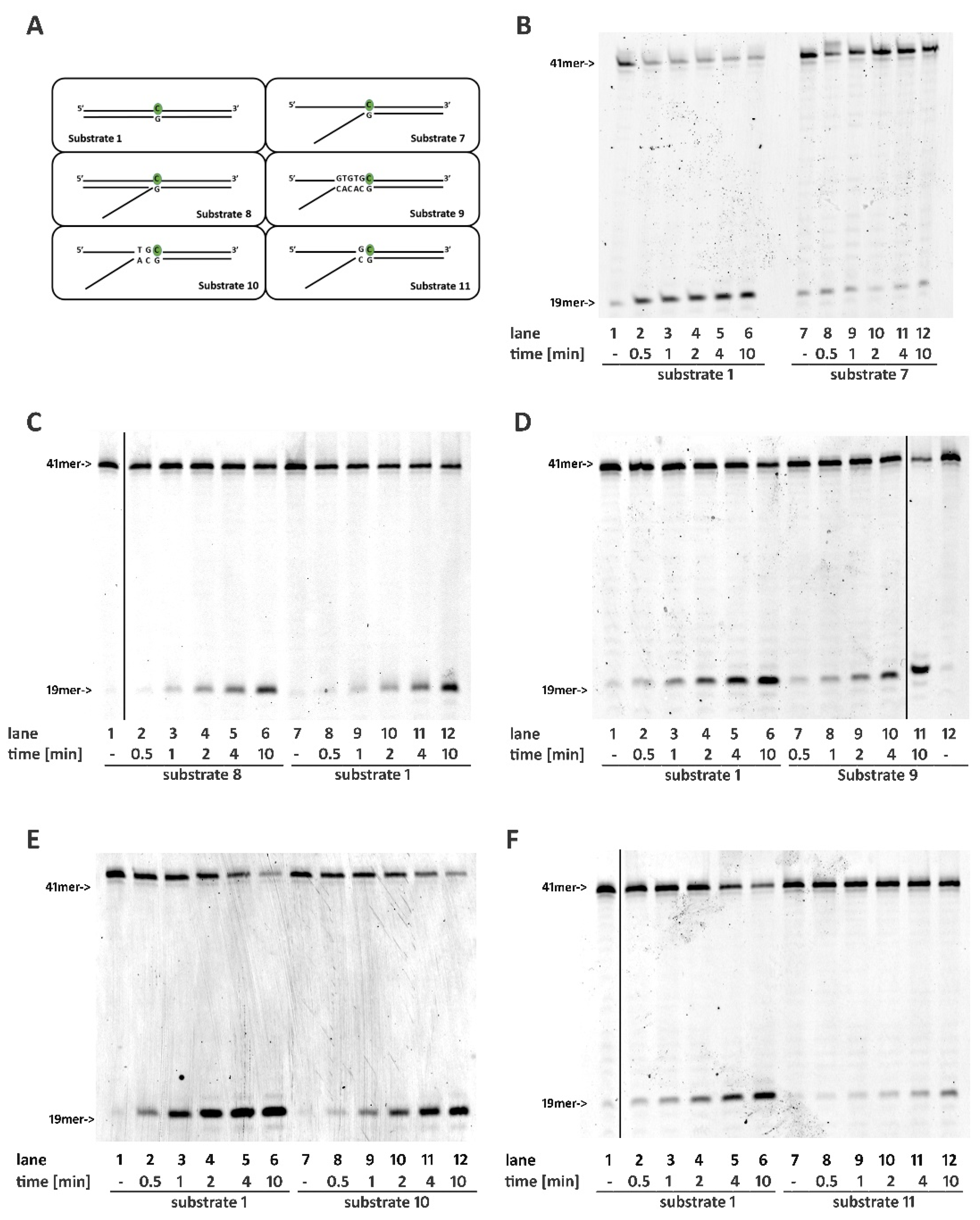
2.3. RNase H2 Can Process Ribonucleotides at the Junction of Flap-Like Structures at the 3′ of the Embedded Ribonucleotide
2.4. Base Pairing at the 5′ of the Embedded Ribonucleotide Is Essential for RNase H2 Activity
2.5. DNA Mismatches at the 5′ of the Ribonucleotide Differentially Affects RNase H2 Activity
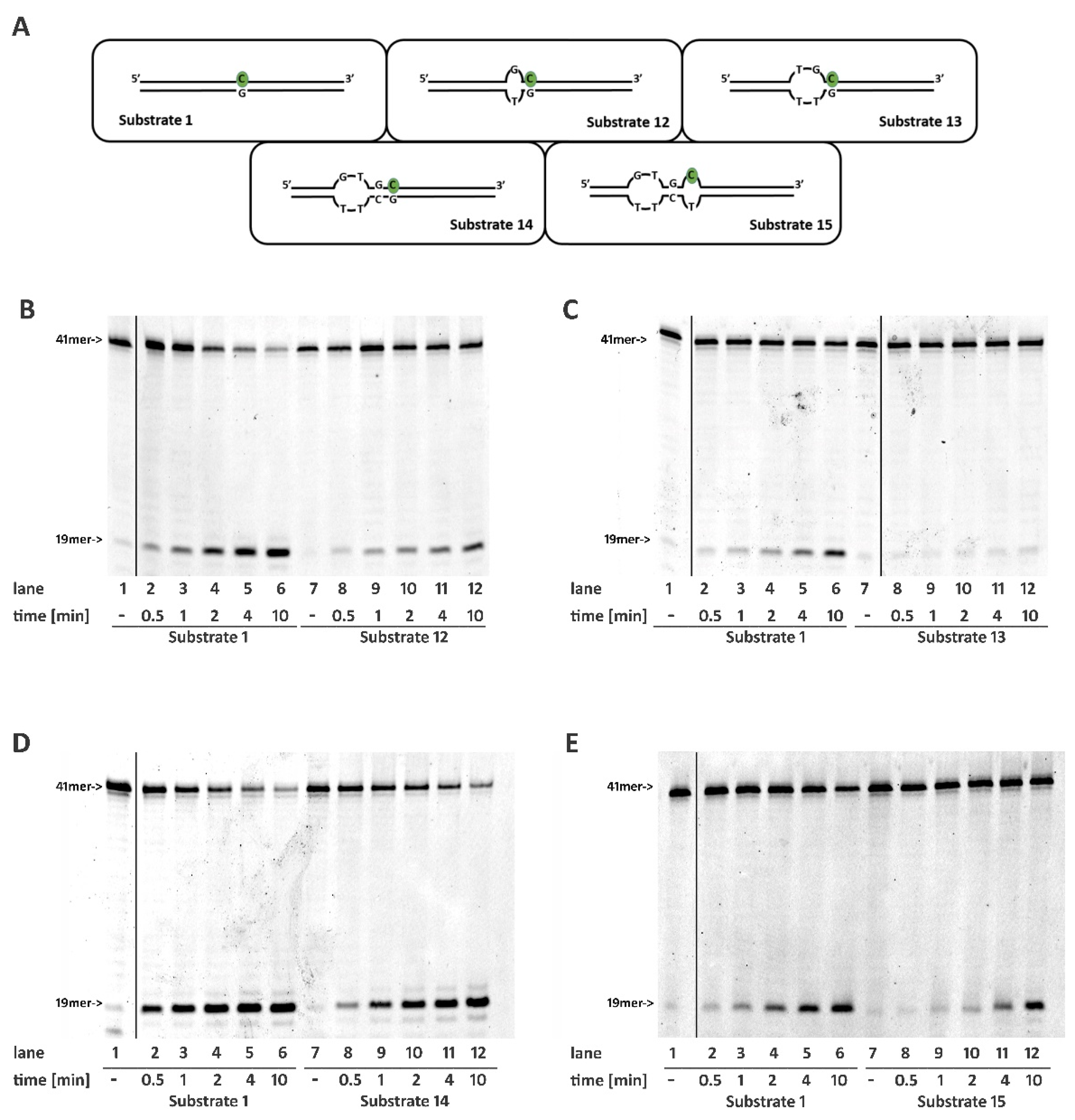
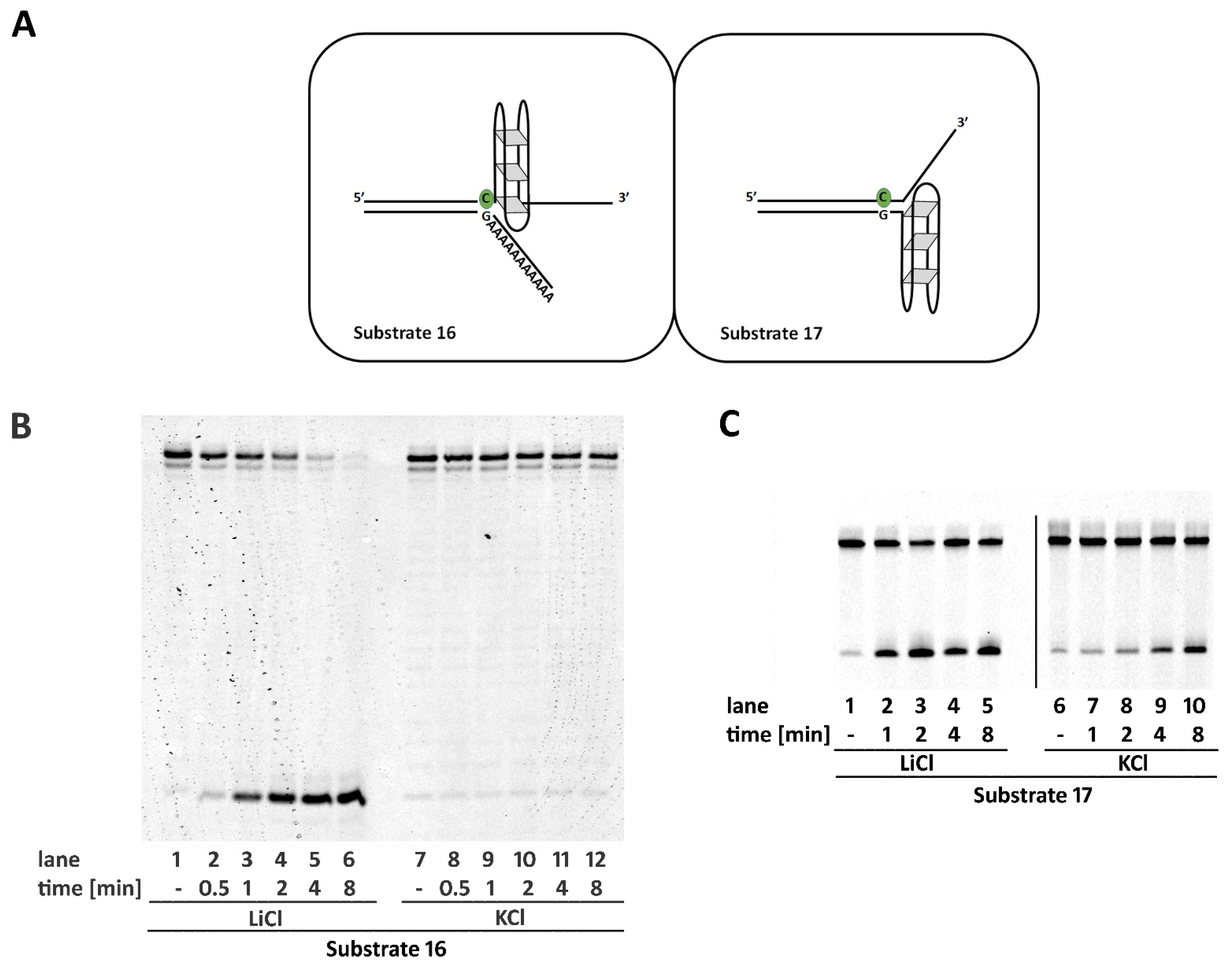
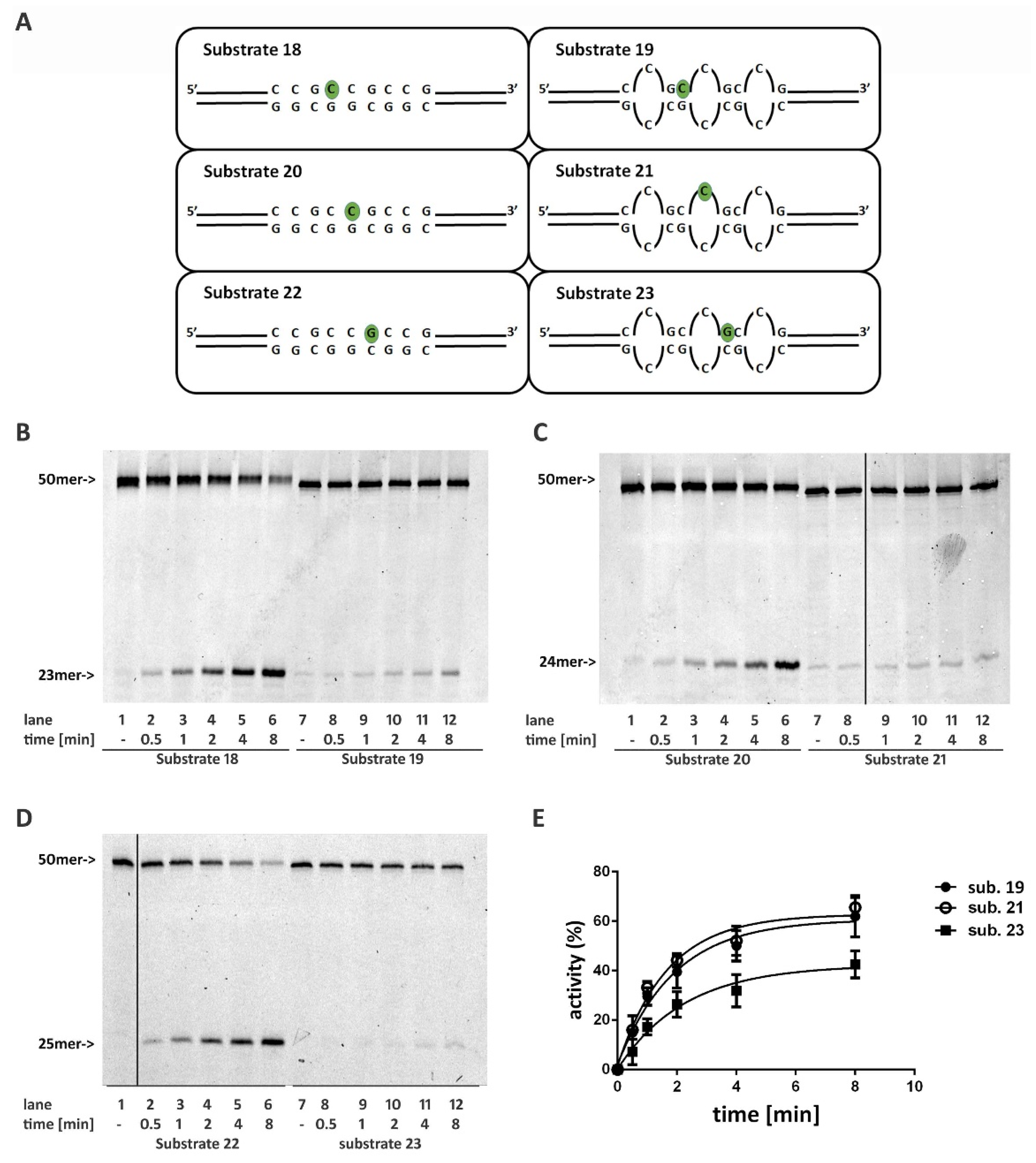
2.6. Non-B DNA Structures Affect RNase H2 Activity
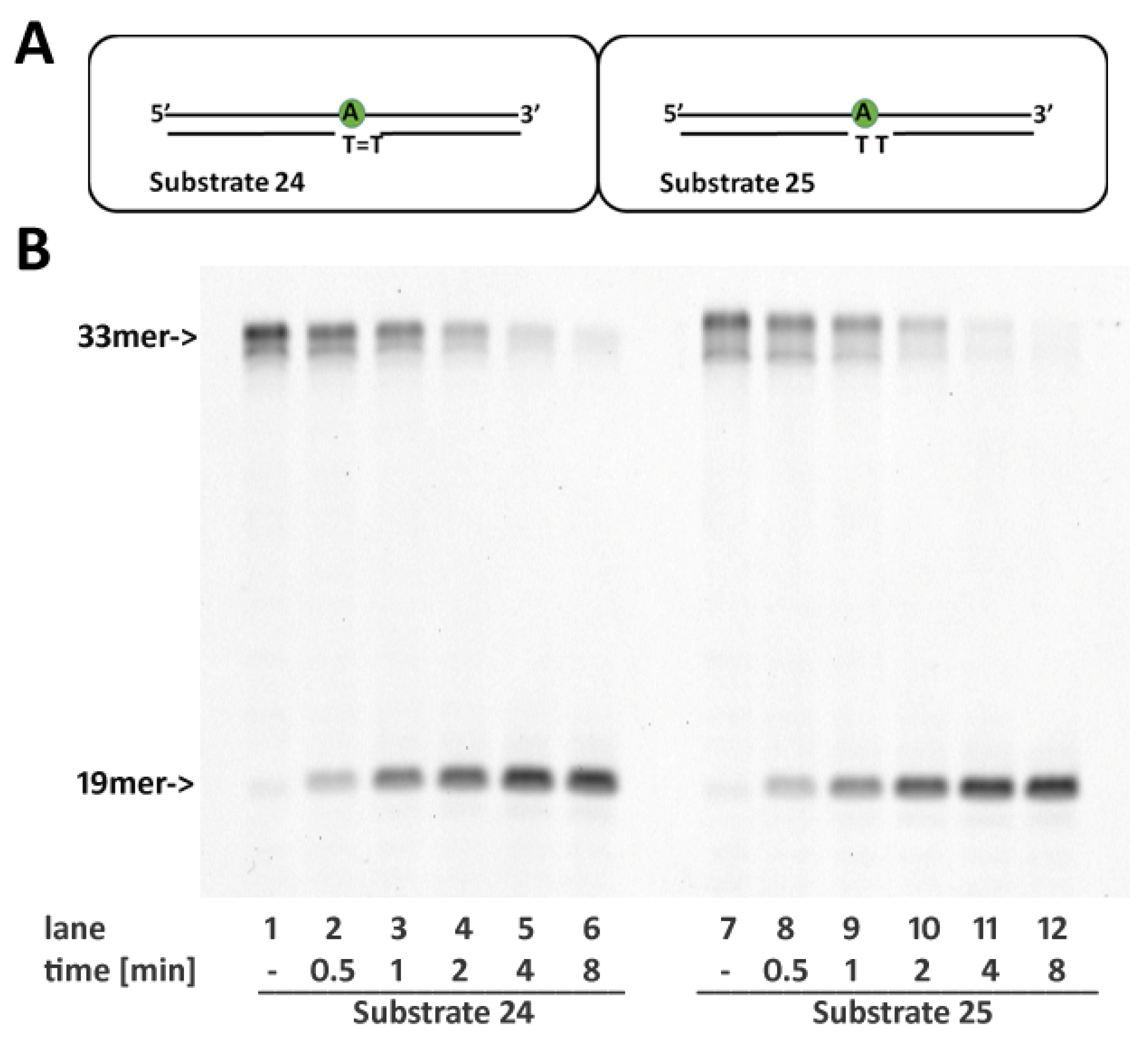
2.7. RNase H2 Activity Is Not Affected by Cyclobutane Thymine Dimers Opposite the Embedded Ribonucleotide
3. Discussion
4. Materials and Methods
4.1. DNA Substrates
4.2. RNase H2 Purification
4.3. RNase H2 Assay and Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caldecott, K.W. Molecular biology. Ribose--An internal threat to DNA. Science 2014, 343, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundström, E.B.; Burgers, P.M.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.Y.; Schroeder, J.W.; Yurieva, O.; Simmons, L.A.; O’Donnell, M.E. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. USA 2013, 110, 12942–12947. [Google Scholar] [CrossRef] [PubMed]
- Ghodgaonkar, M.M.; Lazzaro, F.; Olivera-Pimentel, M.; Artola-Borán, M.; Cejka, P.; Reijns, M.A.; Jackson, A.P.; Plevani, P.; Muzi-Falconi, M.; Jiricny, J. Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol. Cell 2013, 50, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Williams, J.S.; Clausen, A.R.; Clark, A.B.; Kunkel, T.A. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol. Cell 2013, 50, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef]
- Clausen, A.R.; Murray, M.S.; Passer, A.R.; Pedersen, L.C.; Kunkel, T.A. Structure-function analysis of ribonucleotide bypass by B family DNA replicases. Proc. Natl. Acad. Sci. USA 2013, 110, 16802–16807. [Google Scholar] [CrossRef]
- Clausen, A.R.; Zhang, S.; Burgers, P.M.; Lee, M.Y.; Kunkel, T.A. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair 2013, 12, 121–127. [Google Scholar] [CrossRef]
- Watt, D.L.; Johansson, E.; Burgers, P.M.; Kunkel, T.A. Replication of ribonucleotide-containing DNA templates by yeast replicative polymerases. DNA Repair 2011, 10, 897–902. [Google Scholar] [CrossRef]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef]
- Lazzaro, F.; Novarina, D.; Amara, F.; Watt, D.L.; Stone, J.E.; Costanzo, V.; Burgers, P.M.; Kunkel, T.A.; Plevani, P.; Muzi-Falconi, M. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell 2012, 45, 99–110. [Google Scholar] [CrossRef]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef]
- Mentegari, E.; Crespan, E.; Bavagnoli, L.; Kissova, M.; Bertoletti, F.; Sabbioneda, S.; Imhof, R.; Sturla, S.J.; Nilforoushan, A.; Hübscher, U.; et al. Ribonucleotide incorporation by human DNA polymerase η impacts translesion synthesis and RNase H2 activity. Nucleic Acids Res. 2017, 45, 2600–2614. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.S. The human specialized DNA polymerases and non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.Y.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef] [PubMed]
- Del Villar-Guerra, R.; Trent, J.O.; Chaires, J.B. G-Quadruplex Secondary Structure Obtained from Circular Dichroism Spectroscopy. Angew. Chem. 2018, 57, 7171–7175. [Google Scholar] [CrossRef] [PubMed]
- Kypr, J.; Kejnovská, I.; Renciuk, D.; Vorlícková, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Kharel, P.; Becker, G.; Tsvetkov, V.; Ivanov, P. Properties and biological impact of RNA G-quadruplexes: From order to turmoil and back. Nucleic Acids Res. 2020, 48, 12534–12555. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Houlden, H.; Tabrizi, S.J. DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 2017, 16, 88–96. [Google Scholar] [CrossRef]
- Farahani, N.; Behmanesh, M.; Ranjbar, B. Evaluation of Rationally Designed Label-free Stem-loop DNA Probe Opening in the Presence of miR-21 by Circular Dichroism and Fluorescence Techniques. Sci. Rep. 2020, 10, 4018. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Koh, K.D.; Weiss, B.; Storici, F. Mispaired rNMPs in DNA are mutagenic and are targets of mismatch repair and RNases, H. Nat. Struct. Mol. Biol. 2011, 19, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.P.; Chon, H.; Cerritelli, S.M.; Klimek, P.; Crouch, R.J.; Nowotny, M. Crystal structures of RNase H2 in complex with nucleic acid reveal the mechanism of RNA-DNA junction recognition and cleavage. Mol. Cell 2010, 40, 658–670. [Google Scholar] [CrossRef]
- Zheng, L.; Shen, B. Okazaki fragment maturation: Nucleases take centre stage. J. Mol. Cell Biol. 2011, 3, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Renčiuk, D.; Kypr, J.; Vorlíčková, M. CGG repeats associated with fragile X chromosome form left-handed Z-DNA structure. Biopolymers 2011, 95, 174–181. [Google Scholar] [CrossRef]
- Yang, W. An overview of Y-Family DNA polymerases and a case study of human DNA polymerase η. Biochemistry 2014, 53, 2793–2803. [Google Scholar] [CrossRef]
- Su, Y.; Egli, M.; Guengerich, F.P. Mechanism of Ribonucleotide Incorporation by Human DNA Polymerase η. J. Biol. Chem. 2016, 291, 3747–3756. [Google Scholar] [CrossRef]
- Crespan, E.; Furrer, A.; Rösinger, M.; Bertoletti, F.; Mentegari, E.; Chiapparini, G.; Imhof, R.; Ziegler, N.; Sturla, S.J.; Hübscher, U.; et al. Impact of ribonucleotide incorporation by DNA polymerases β and λ on oxidative base excision repair. Nat. Commun. 2016, 7, 10805. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Balachander, S.; Antoniali, G.; Koh, K.D.; Saint-Pierre, C.; Gasparutto, D.; Chon, H.; Crouch, R.J.; Storici, F.; Tell, G. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res. 2017, 45, 11193–11212. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Henneke, G.; Balachander, S.; Koh, K.D.; Newnam, G.; Uehara, R.; Crouch, R.J.; Storici, F.; Tell, G. Unlike the Escherichia coli counterpart, archaeal RNase HII cannot process ribose monophosphate abasic sites and oxidized ribonucleotides embedded in DNA. J. Biol. Chem. 2019, 294, 13061–13072. [Google Scholar] [CrossRef]
- Vaisman, A.; McDonald, J.P.; Huston, D.; Kuban, W.; Liu, L.; Van Houten, B.; Woodgate, R. Removal of misincorporated ribonucleotides from prokaryotic genomes: An unexpected role for nucleotide excision repair. PLoS Genet. 2013, 9, e1. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Oligo | Sequence |
|---|---|---|
| 1 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #2 | 5′-TATCCACCAATACTACCCTACGCACACCTCCACTCAAACTC-3′ | |
| 2 | #3 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #4 | 5′-TGAGGAGTTGGTTGCACACCTCCACTCAAACTC-3′ | |
| 3 | #1 | Fam |
| #5 | 5′-TGAGGAGTTGGTTTCACACCTCCACTCAAACTC-3′ | |
| 4 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #6 | 5′-TGAGGAGTTGGTTTCACACCTCCACTCAAACTC-3′ | |
| 5 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #5 | 5′-TGAGGAGTTGGTTGCACACCTCCACTCAAACTC-3′ | |
| #7 | 5′-TATCCACCAATACTACCCTAC-3′ | |
| 6 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #6 | 5′-TGAGGAGTTGGTTTCACACCTCCACTCAAACTC-3′ | |
| #7 | 5′-TATCCACCAATACTACCCTAC-3′ | |
| 7 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #8 | 5′-TATCCACCAATACTACCCTACGTTTTGGATGTGGGTTT-3′ | |
| 8 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #8 | 5′-TATCCACCAATACTACCCTACGTTTTGGATGTGGGTTT-3′ | |
| #9 | 5′-CACACCTCCACTCAAACTC-3′ | |
| 9 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #10 | 5′-TATCCACCAATACTACCCTACGTTTTGGATGTGGGTTT-3′ | |
| 10 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #11 | 5′-TATCCACCAATACTACCCTACGCTTTGGATGTGGGTTT-3′ | |
| 11 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #12 | 5′-TATCCACCAATACTACCCTACGCATTGGATGTGGGTTT-3′ | |
| 12 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #13 | 5′-TATCCACCAATACTACCCTACGTACACCTCCACTCAAACTC-3′ | |
| 13 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #14 | 5′-TATCCACCAATACTACCCTACGTTCACCTCCACTCAAACTC-3′ | |
| 14 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #15 | 5′-TATCCACCAATACTACCCTACGCTGACCTCCACTCAAACTC-3′ | |
| 15 | #1 | 5′-Fam-GAGTTTGAGTGGAGGTGTG(rC)GTAGGGTAGTATTGGTGGATA-3′ |
| #16 | 5′-TATCCACCAATACTACCCTACTCTGACCTCCACTCAAACTC-3′ | |
| 16 | #17 | 5′-Fam-GAGAGAAGATGGAAGTGAGGAA(rC)GGGATAGGGATAGGGATAGGG |
| #18 | 5′-AAAAAAAAAAAAAAAAAAAAAGTTCCTCACTTCCATCTTCTCTC-3′ | |
| 17 | #19 | 5′-Fam-GAGAGAAGATGGAAGTGAGGAA(rC)AAAAAAAAAAAAAAAAAAAAAAA-3′ |
| #20 | 5′-GGGATAGGGATAGGGATAGGGTTGTTCCTCACTTCCATCTTCTCTC-3′ | |
| 18 | #21 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCG(rC)CGCCGGTAGAGTAGTATGTGTTGATA-3′ |
| #22 | 5′-TATCAACACATACTACTCTACGCCGCCGCCACAACACTACACTCAAACTC-3′ | |
| 19 | #21 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCG(rC)CGCCGGTAGAGTAGTATGTGTTGATA-3′ |
| #23 | 5′-TATCAACACATACTACTCTACCGGCGGCGGACAACACTACACTCAAAC-3′ | |
| 20 | #24 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCGC(rC)GCCGGTAGAGTAGTATGTGTTGATA-3′ |
| #22 | 5′-TATCAACACATACTACTCTACGCCGCCGCCACAACACTACACTCAAACTC-3′ | |
| 21 | #24 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCGC(rC)GCCGGTAGAGTAGTATGTGTTGATA-3′ |
| #23 | 5′-TATCAACACATACTACTCTACCGGCGGCGGACAACACTACACTCAAAC-3′ | |
| 22 | #25 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCGCC(rG)CCGGTAGAGTAGTATGTGTTGATA-3′ |
| #22 | 5′-TATCAACACATACTACTCTACGCCGCCGCCACAACACTACACTCAAACTC-3′ | |
| 23 | #25 | 5′-Fam-GAGTTTGAGTGTAGTGTTGTCCGCC(rG)CCGGTAGAGTAGTATGTGTTGATA-3′ |
| #23 | 5′-TATCAACACATACTACTCTACCGGCGGCGGACAACACTACACTCAAAC-3′ | |
| 24 | #26 | 5′-Fam-GGGTGGAGGTGACA[rA]GAGTGTGATAGAGTGTGA-3′ |
| #27 | 5′-TCACACTCTATCACACTC[CPD]GTCACCTCCACCC | |
| 25 | #26 | 5′-Fam-GGGTGGAGGTGACA[rA]GAGTGTGATAGAGTGTGA-3′ |
| #28 | 5′-TCACACTCTATCACACTCTTGTCACCTCCACCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dede, M.; Napolitano, S.; Melati, A.; Pirota, V.; Maga, G.; Crespan, E. High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures. Int. J. Mol. Sci. 2021, 22, 5201. https://doi.org/10.3390/ijms22105201
Dede M, Napolitano S, Melati A, Pirota V, Maga G, Crespan E. High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures. International Journal of Molecular Sciences. 2021; 22(10):5201. https://doi.org/10.3390/ijms22105201
Chicago/Turabian StyleDede, Maria, Silvia Napolitano, Anna Melati, Valentina Pirota, Giovanni Maga, and Emmanuele Crespan. 2021. "High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures" International Journal of Molecular Sciences 22, no. 10: 5201. https://doi.org/10.3390/ijms22105201
APA StyleDede, M., Napolitano, S., Melati, A., Pirota, V., Maga, G., & Crespan, E. (2021). High Flexibility of RNaseH2 Catalytic Activity with Respect to Non-Canonical DNA Structures. International Journal of Molecular Sciences, 22(10), 5201. https://doi.org/10.3390/ijms22105201