
MiR-15a-5p Confers Chemoresistance in Acute Myeloid Leukemia by Inhibiting Autophagy Induced by Daunorubicin

 and
and
Abstract
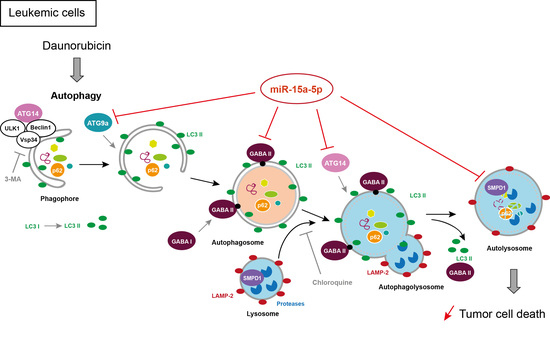
1. Introduction
2. Results
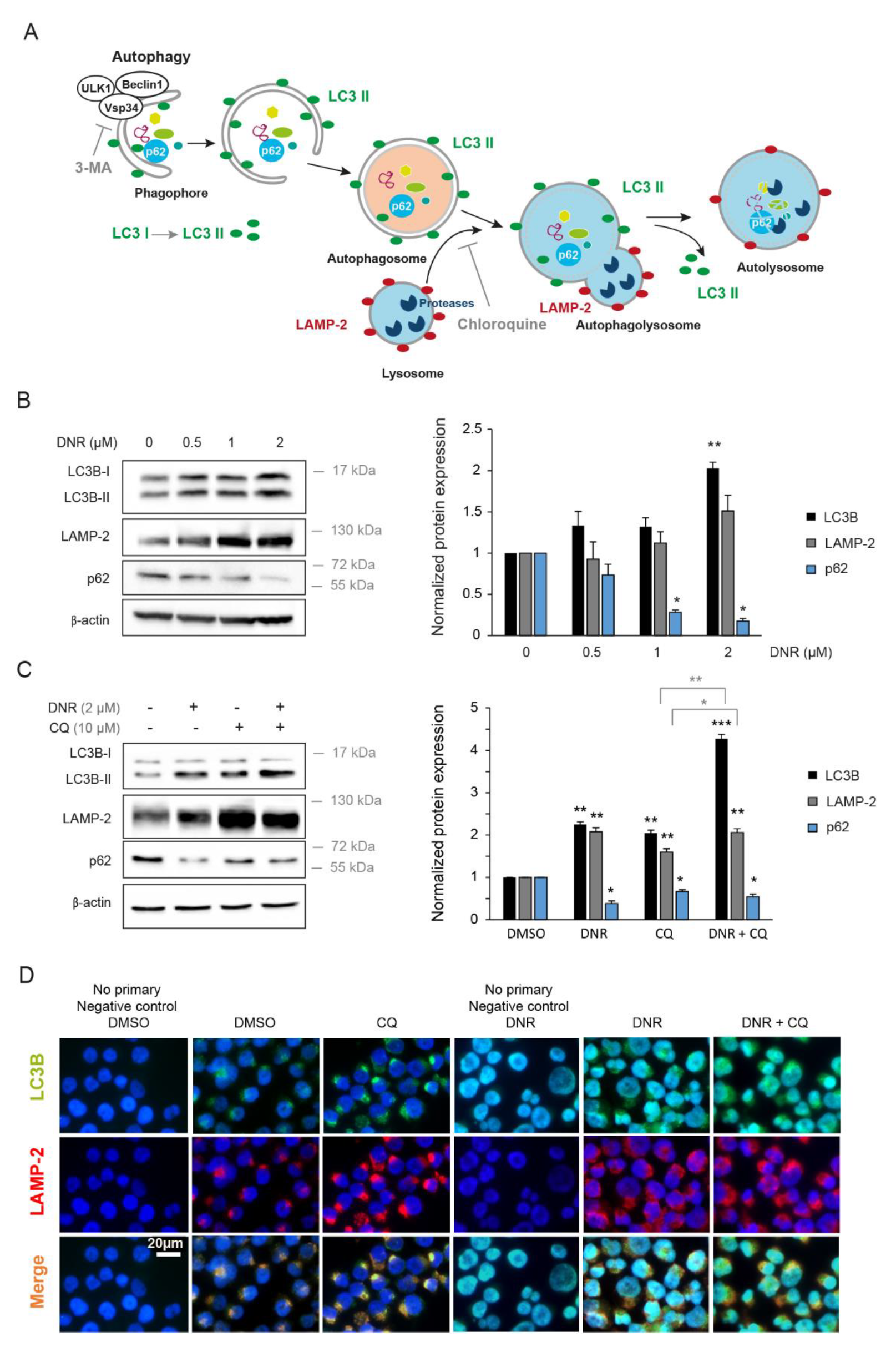
2.1. Daunorubicin Induces Autophagy in Myeloid Cell Lines
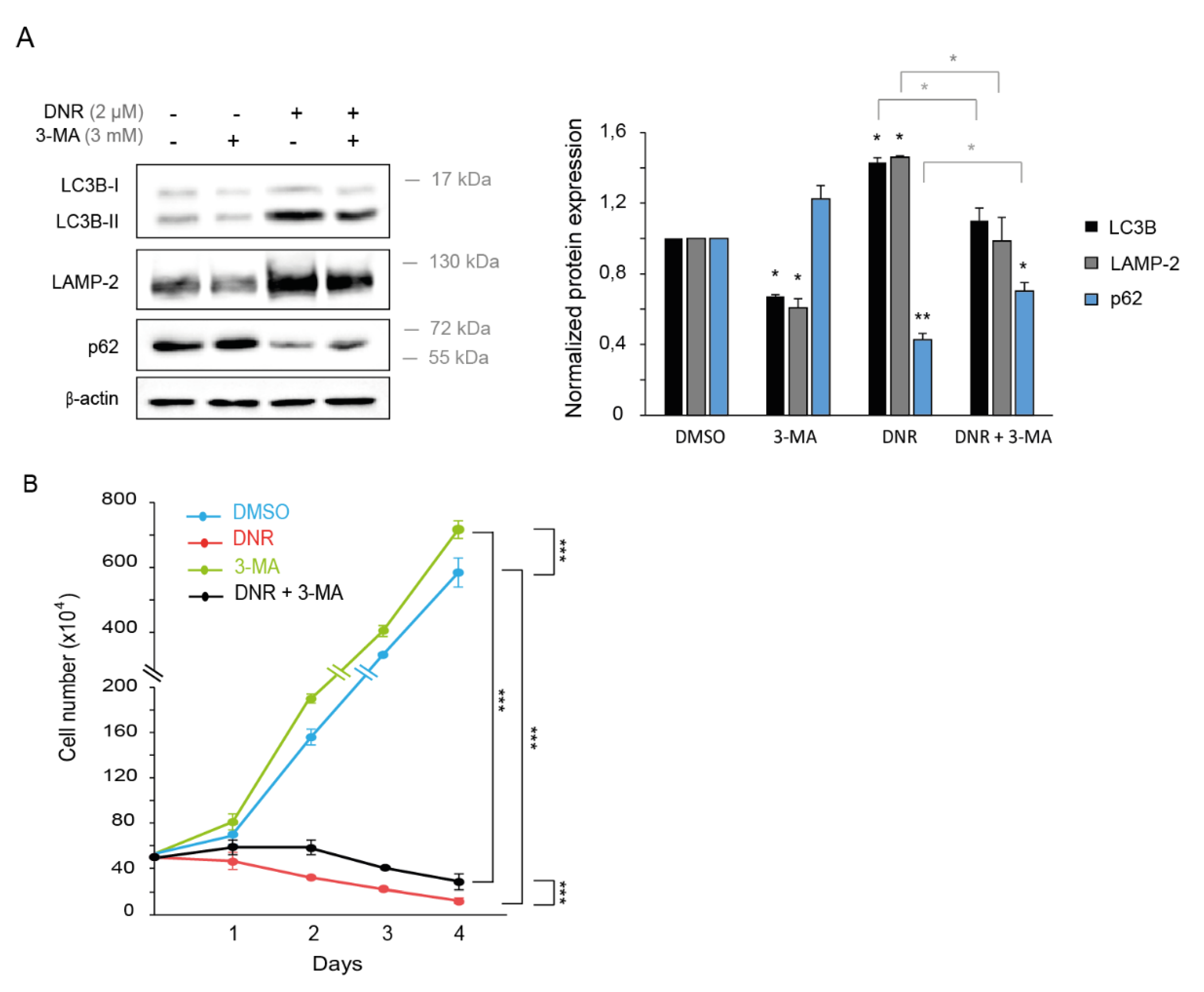
2.2. Autophagy Inhibition Increases Myeloid Cell Growth and Prevents Downregulation by Daunorubicin
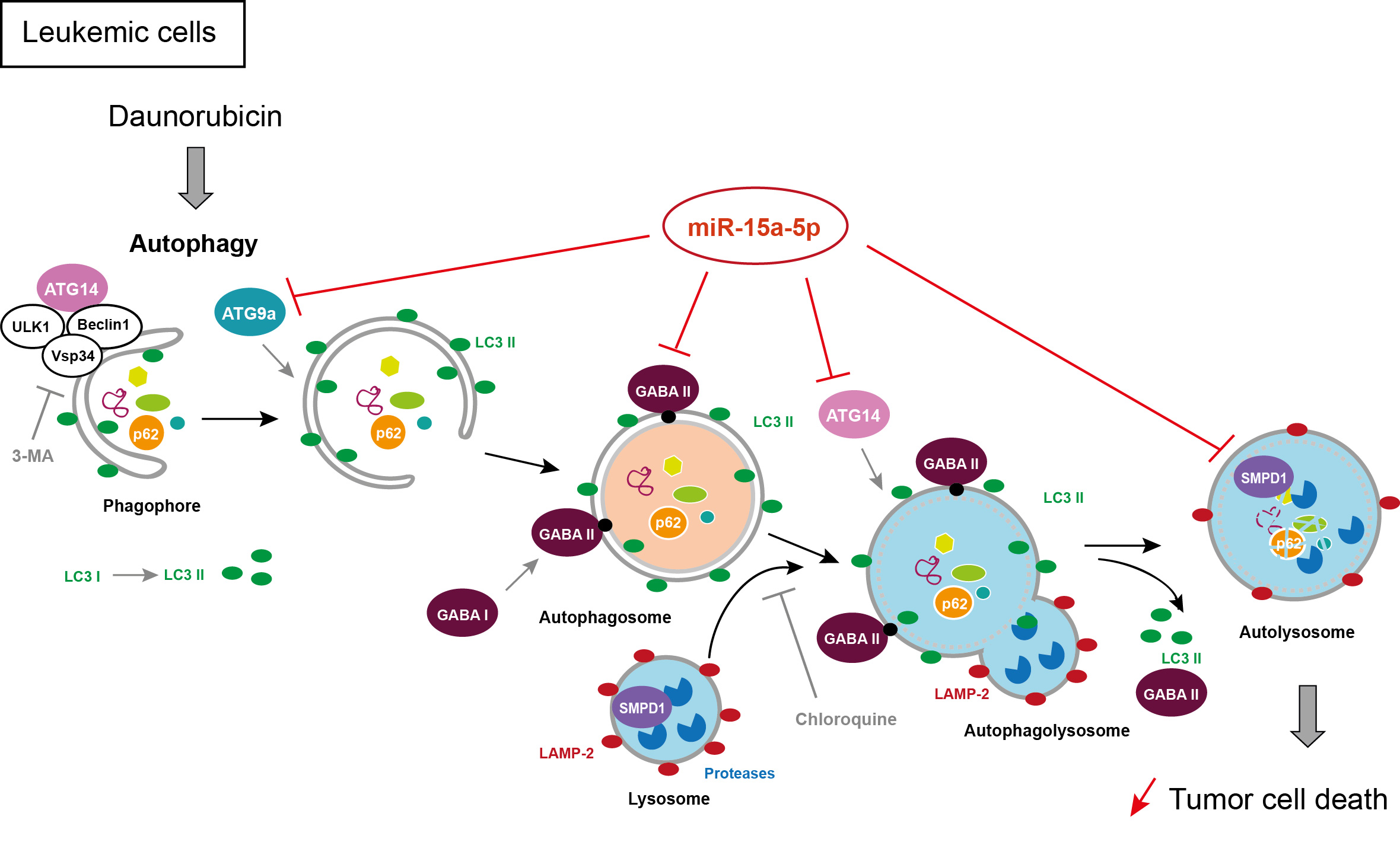
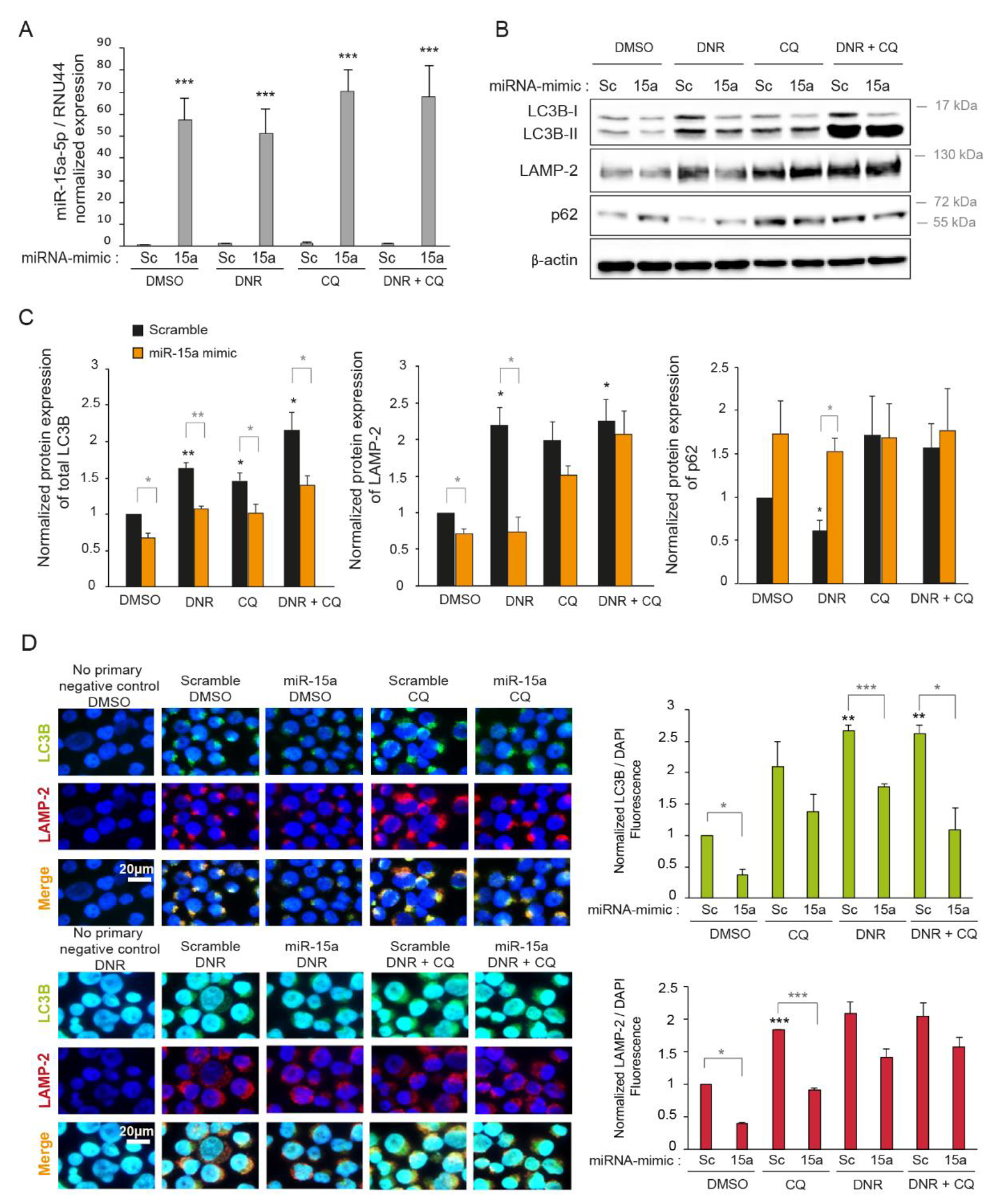
2.3. Overexpression of miR-15a-5p Inhibits Autophagy Induced by Daunorubicin
2.4. Overexpression of miR-15a-5p Increases Myeloid Cell Growth and Prevents Downregulation by Daunorubicin
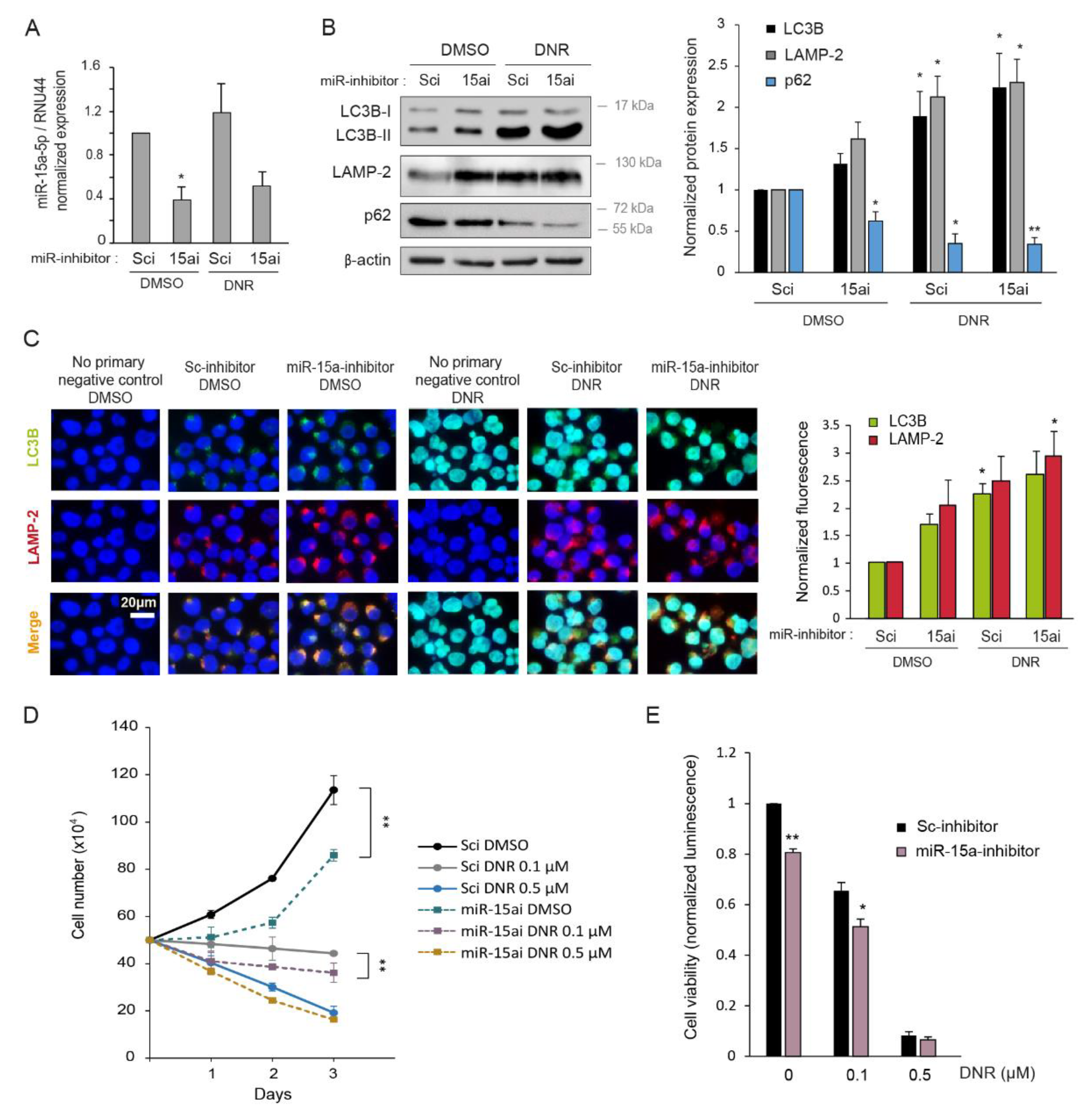
2.5. Inhibition of miR-15a-5p Induces Autophagy and Decreases Myeloid Cell Growth
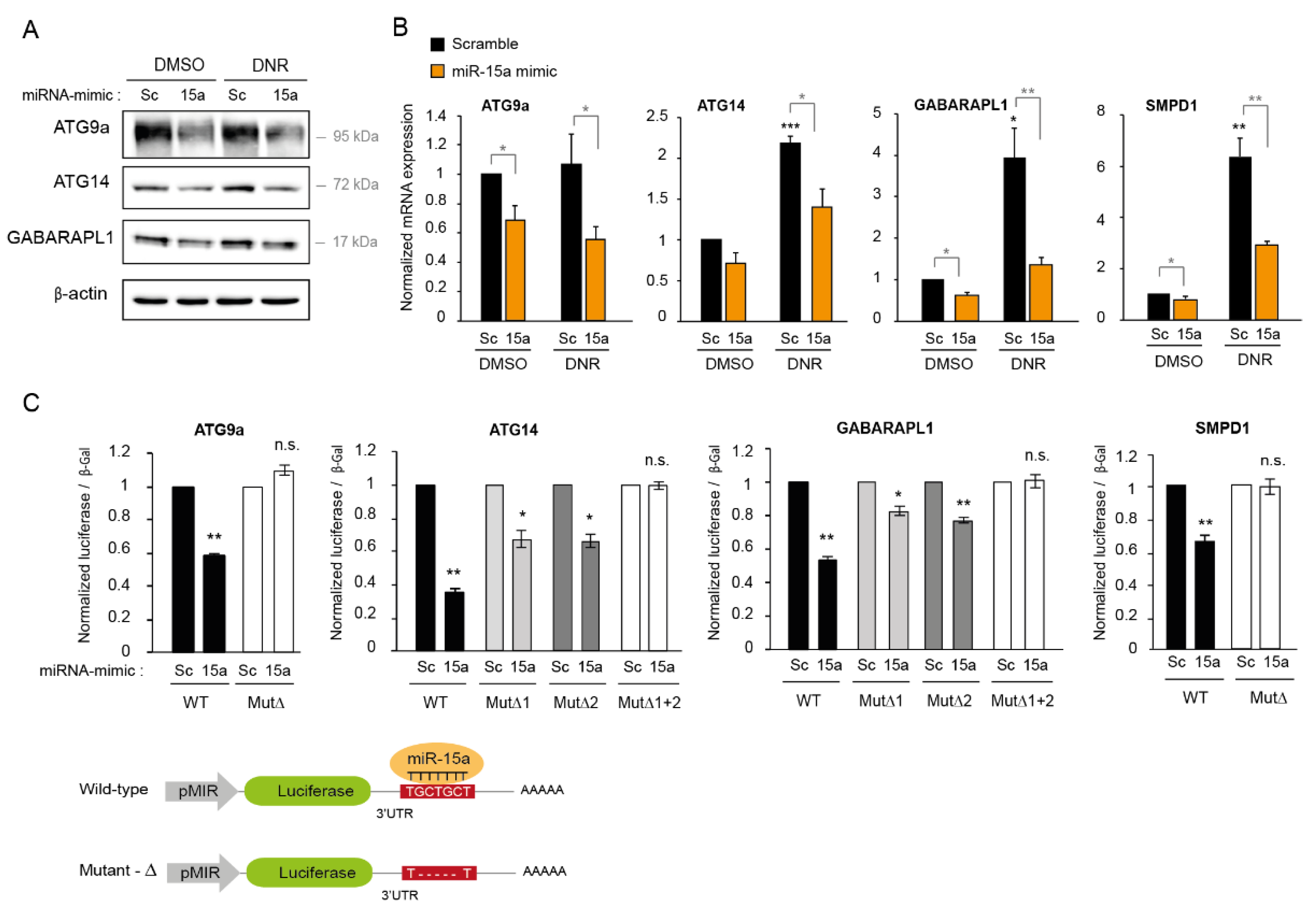
2.6. MiR-15a-5p Downregulates the Expression of Autophagy Target Genes
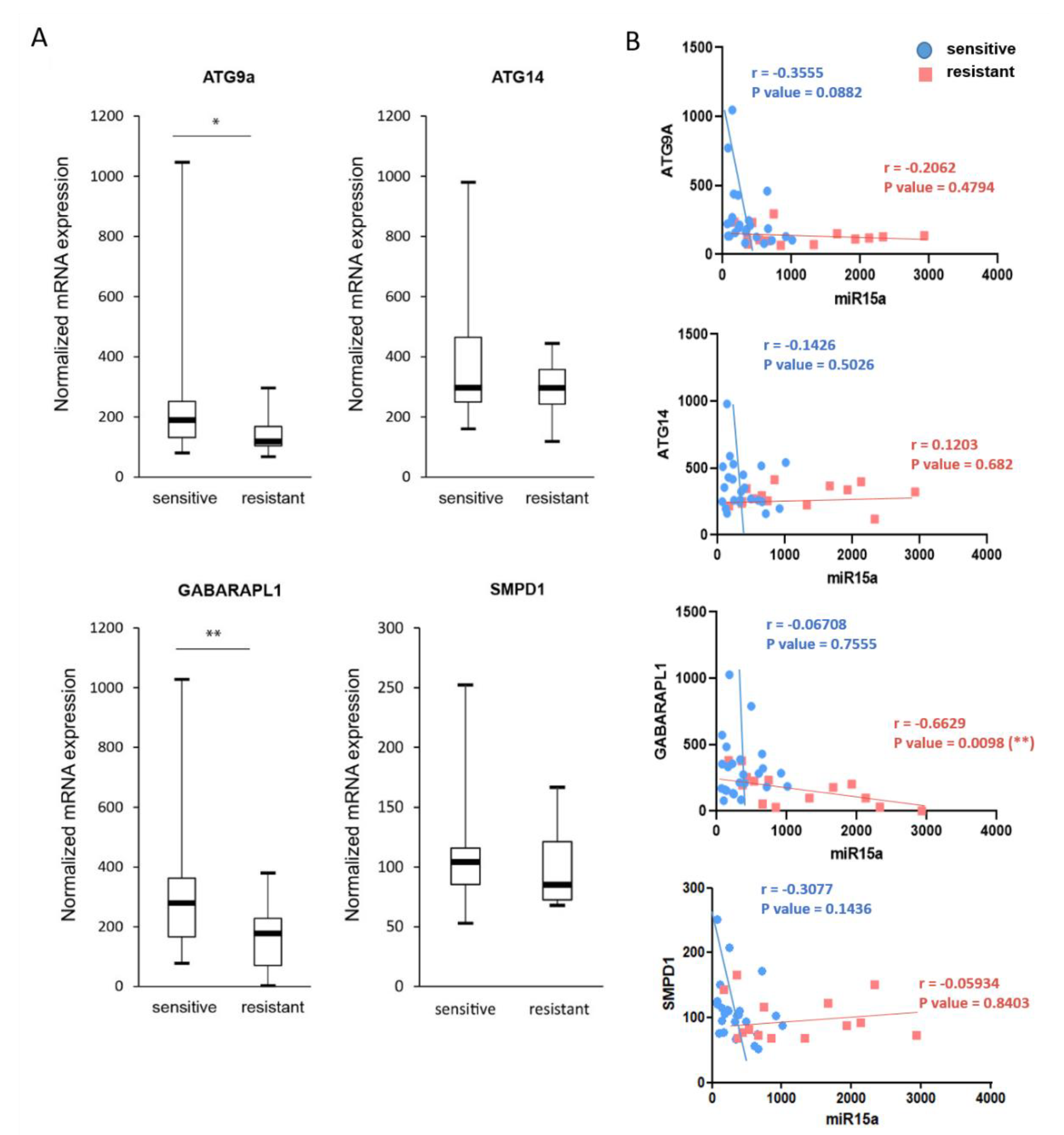
2.7. Expression of ATG9A, ATG14, and GABARAPL1 Are Downregulated in Chemoresistant AML Patients
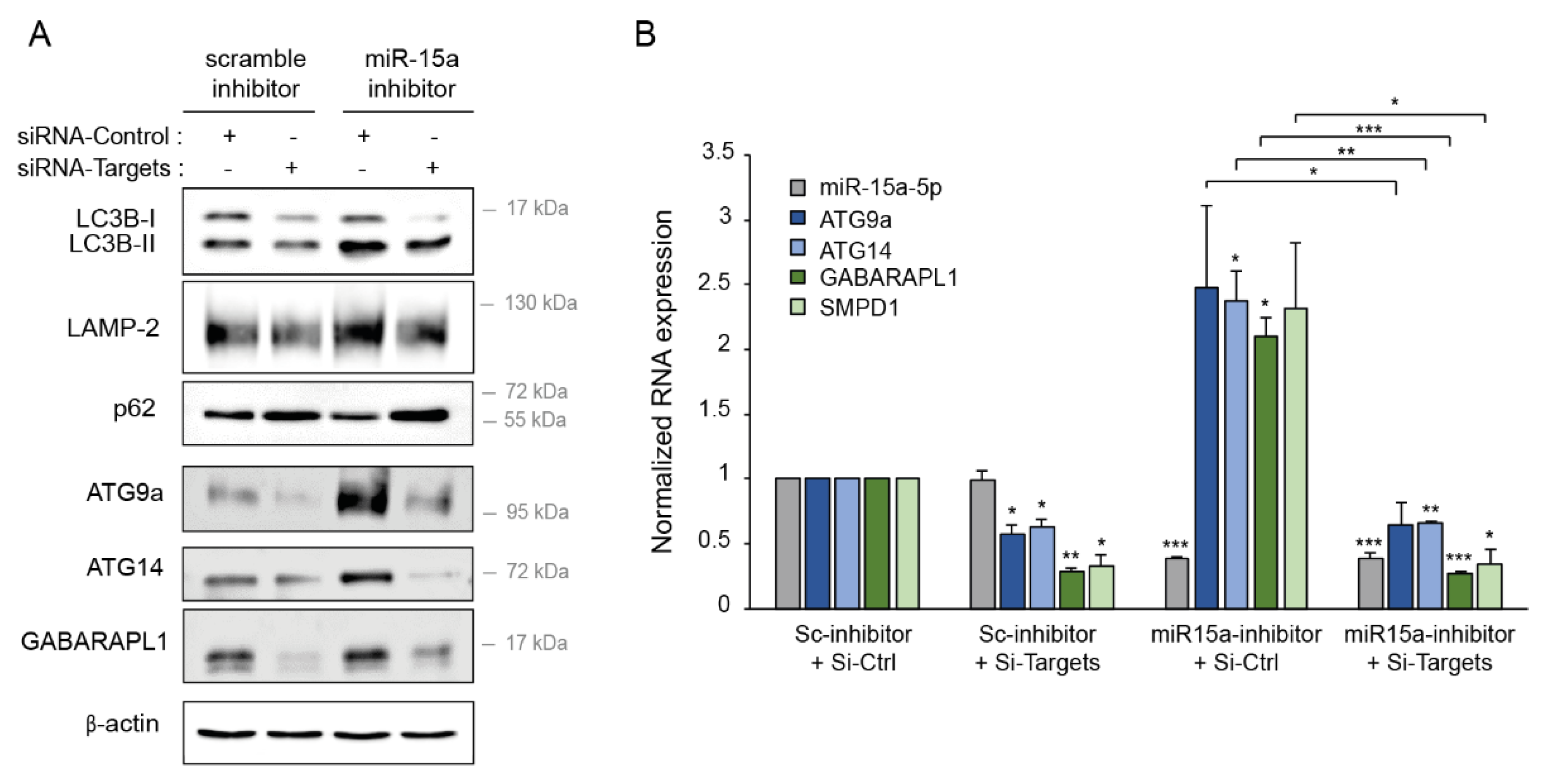
2.8. Downregulation of miR-15a-5p Targets Decreases the Activation of Autophagy
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Cell Transient Transfection
4.4. Protein Extraction and Western Blotting
4.5. RNA Extraction and RT-qPCR
4.6. Immunofluorescence
4.7. Cell viability Assay
4.8. Microarray Assay
4.9. Cloning and Site-Directed Mutagenesis
4.10. Luciferase Assays
4.11. Gene Expression Profiles of Patient Samples
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dinmohamed, A.G.; Visser, O.; van Norden, Y.; Blijlevens, N.M.; Cornelissen, J.J.; Huls, G.A.; Huijgens, P.C.; Sonneveld, P.; van de Loosdrecht, A.A.; Ossenkoppele, G.J.; et al. Treatment, trial participation and survival in adult acute myeloid leukemia: A population-based study in the Netherlands, 1989–2012. Leukemia 2016, 30, 24–31. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Chan, S.M.; Daver, N.G.; Jonas, B.A.; Pollyea, D.A. Optimizing survival outcomes with post-remission therapy in acute myeloid leukemia. Am. J. Hematol. 2019, 94, 803–811. [Google Scholar] [CrossRef]
- Lowenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef]
- Luskin, M.R.; Lee, J.W.; Fernandez, H.F.; Abdel-Wahab, O.; Bennett, J.M.; Ketterling, R.P.; Lazarus, H.M.; Levine, R.L.; Litzow, M.R.; Paietta, E.M.; et al. Benefit of high-dose daunorubicin in AML induction extends across cytogenetic and molecular groups. Blood 2016, 127, 1551–1558. [Google Scholar] [CrossRef]
- Gong, Q.; Zhou, L.; Xu, S.; Li, X.; Zou, Y.; Chen, J. High Doses of Daunorubicin during Induction Therapy of Newly Diagnosed Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis of Prospective Clinical Trials. PLoS ONE 2015, 10, e0125612. [Google Scholar] [CrossRef]
- Isakson, P.; Bjoras, M.; Boe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Jin, J.; Britschgi, A.; Schlafli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef] [PubMed]
- Havelange, V.; Garzon, R.; Croce, C.M. MicroRNAs: New players in acute myeloid leukaemia. Br. J. Cancer 2009, 101, 743–748. [Google Scholar] [CrossRef]
- Marcucci, G.; Mrozek, K.; Radmacher, M.D.; Garzon, R.; Bloomfield, C.D. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood 2011, 117, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Akkoc, Y.; Ozturk, D.G.; Kocak, M. Autophagy-Regulating microRNAs and Cancer. Front. Oncol. 2017, 7, 65. [Google Scholar] [CrossRef]
- Kovaleva, V.; Mora, R.; Park, Y.J.; Plass, C.; Chiramel, A.I.; Bartenschlager, R.; Dohner, H.; Stilgenbauer, S.; Pscherer, A.; Lichter, P.; et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012, 72, 1763–1772. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Gabra, M.M.; Salmena, L. microRNAs and Acute Myeloid Leukemia Chemoresistance: A Mechanistic Overview. Front. Oncol. 2017, 7, 255. [Google Scholar] [CrossRef]
- Zebisch, A.; Hatzl, S.; Pichler, M.; Wolfler, A.; Sill, H. Therapeutic Resistance in Acute Myeloid Leukemia: The Role of Non-Coding RNAs. Int. J. Mol. Sci. 2016, 17, 2080. [Google Scholar] [CrossRef]
- Li, H.; Hui, L.; Xu, W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Acta Biochim. Biophys. Sin. 2012, 44, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Cao, Z.; Deng, C.; Zhou, L.; Wang, C. miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C by inducing apoptosis. J. Cancer Res. Clin. Oncol. 2012, 138, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, V.; Essaghir, A.; Bollaert, E.; Lenglez, S.; Graux, C.; Schoemans, H.; Saussoy, P.; Michaux, L.; Valk, P.J.M.; Demoulin, J.; et al. miR-15a-5p and miR-21-5p contribute to chemoresistance in cytogenetically normal acute myeloid leukemia by targeting PDCD4, ARL2 and BTG2. J. Cell. Mol. Med. 2021, 25, 575–585. [Google Scholar] [CrossRef]
- Butrym, A.; Baczynska, D.; Tukiendorf, A.; Rybka, J.; Dobosz, T.; Jurczak, W.; Kuliczkowski, K.; Mazur, G. High Expression of Mir-15a Predicts Shorter Survival and Worse Response to Chemotherapy in Patients with Acute Myeloid Leukemia (AML). Blood 2014, 124, 5530. [Google Scholar] [CrossRef]
- Jing, K.; Lim, K. Why is autophagy important in human diseases? Exp. Mol. Med. 2012, 44, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Sun, J.; Feng, L.; Wang, K.; Li, D.; Pan, Q.; Chen, Y.; Jin, W.; Wang, X.; Pan, H.; et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS ONE 2011, 6, e28491. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Chakrama, F.Z.; Seguin-Py, S.; Le Grand, J.N.; Fraichard, A.; Delage-Mourroux, R.; Despouy, G.; Perez, V.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010, 6, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, M.; Pitzer, A.L.; Xia, M.; Boini, K.M.; Li, P.L.; Zhang, Y. Control of autophagy maturation by acid sphingomyelinase in mouse coronary arterial smooth muscle cells: Protective role in atherosclerosis. J. Mol. Med. 2014, 92, 473–485. [Google Scholar] [CrossRef]
- Valk, P.J.; Verhaak, R.G.; Beijen, M.A.; Erpelinck, C.A.; van Doorn-Khosrovani, S.B.V.W.; Boer, J.M.; Beverloo, H.B.; Moorhouse, M.J.; van der Spek, P.J.; Lowenberg, B.; et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1617–1628. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Ristic, B.; Bosnjak, M.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Bogdanovic, A.; Perovic, V.; Martinovic, T.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Idarubicin induces mTOR-dependent cytotoxic autophagy in leukemic cells. Exp. Cell Res. 2014, 326, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.P.; Niu, Y.N.; Yuan, N.; Zhang, A.H.; Chao, D.; Xu, Q.P.; Wang, L.J.; Zhang, X.G.; Zhao, W.L.; Zhao, Y.; et al. Role of autophagy in acute myeloid leukemia therapy. Chin. J. Cancer 2013, 32, 130–135. [Google Scholar] [CrossRef]
- Seca, H.; Lima, R.T.; Lopes-Rodrigues, V.; Guimaraes, J.E.; Almeida, G.M.; Vasconcelos, M.H. Targeting miR-21 induces autophagy and chemosensitivity of leukemia cells. Curr. Drug Targets 2013, 14, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ren, W.; Chen, K. MiR-34a Promotes Apoptosis and Inhibits Autophagy by Targeting HMGB1 in Acute Myeloid Leukemia Cells. Cell. Physiol. Biochem. 2017, 41, 1981–1992. [Google Scholar] [CrossRef]
- Zeng, C.W.; Chen, Z.H.; Zhang, X.J.; Han, B.W.; Lin, K.Y.; Li, X.J.; Wei, P.P.; Zhang, H.; Li, Y.; Chen, Y.Q. MIR125B1 represses the degradation of the PML-RARA oncoprotein by an autophagy-lysosomal pathway in acute promyelocytic leukemia. Autophagy 2014, 10, 1726–1737. [Google Scholar] [CrossRef]
- Wallace, J.A.; O’Connell, R.M. MicroRNAs and acute myeloid leukemia: Therapeutic implications and emerging concepts. Blood 2017, 130, 1290–1301. [Google Scholar] [CrossRef]
- Demoulin, J.B.; Ericsson, J.; Kallin, A.; Rorsman, C.; Ronnstrand, L.; Heldin, C.H. Platelet-derived growth factor stimulates membrane lipid synthesis through activation of phosphatidylinositol 3-kinase and sterol regulatory element-binding proteins. J. Biol. Chem. 2004, 279, 35392–35402. [Google Scholar] [CrossRef]
- Essaghir, A.; Dif, N.; Marbehant, C.Y.; Coffer, P.J.; Demoulin, J.B. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J. Biol. Chem. 2009, 284, 10334–10342. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Bollaert, E.; Johanns, M.; Herinckx, G.; de Rocca Serra, A.; Vandewalle, V.A.; Havelange, V.; Rider, M.H.; Vertommen, D.; Demoulin, J.B. HBP1 phosphorylation by AKT regulates its transcriptional activity and glioblastoma cell proliferation. Cell. Signal. 2018, 44, 158–170. [Google Scholar] [CrossRef]
- Velghe, A.I.; Van Cauwenberghe, S.; Polyansky, A.A.; Chand, D.; Montano-Almendras, C.P.; Charni, S.; Hallberg, B.; Essaghir, A.; Demoulin, J.B. PDGFRA alterations in cancer: Characterization of a gain-of-function V536E transmembrane mutant as well as loss-of-function and passenger mutations. Oncogene 2014, 33, 2568–2576. [Google Scholar] [CrossRef]
- Arts, F.A.; Velghe, A.I.; Stevens, M.; Renauld, J.C.; Essaghir, A.; Demoulin, J.B. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J. Cell. Mol. Med. 2015, 19, 239–248. [Google Scholar] [CrossRef]
- Athar, A.; Fullgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Oligonucleotide Forward | Oligonucleotide Reverse |
|---|---|---|
| ATG9A | CTCTGCTAGCTATCCCTGTGC | GCACTGTGCCAGGATCTGT |
| ATG14 | GCGATGAAGAAACCGACCT | CACAAAACCGGGGACTAGG |
| GABARAPL1 | TGGGCCAACTGTATGAGGA | CTACCCCAAGTCCAGGTG |
| SMPD1 | TGGCTCTATGAAGCGATGG | TGGGGAAAGAGCATAGAACC |
| RPLP0 | TCGACAATGGCAGCATCTAC | ATCCGTCTCCACAGACAAGG |
| Mutant | Oligonucleotide |
|---|---|
| ATG9A∆ | CCTGGGCCCTCATTTTATCGTACCCCCC |
| ATG14∆1 | GGGGTGAGTTACACGTATTTTATTCATTCTGTCGTAGTTTGTCAG |
| ATG14∆2 | TAAGTATACATTTCAACCACTGTTTTTTCTACTCTTTTTTCA-TTAAAATCTTTCATGTA |
| GABARAPL1∆1 | TTTCACATGCTCAATTGATATTTTTTTTCCTCGGCCCAGG |
| GABARAPL1∆2 | AGGATTCTTGCTCCCATTGTCCCTTCAGGCTC |
| SMPD1∆ | TCAGGTCGCAAGTACAGGGTTCCTGGCTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bollaert, E.; Claus, M.; Vandewalle, V.; Lenglez, S.; Essaghir, A.; Demoulin, J.-B.; Havelange, V. MiR-15a-5p Confers Chemoresistance in Acute Myeloid Leukemia by Inhibiting Autophagy Induced by Daunorubicin. Int. J. Mol. Sci. 2021, 22, 5153. https://doi.org/10.3390/ijms22105153
Bollaert E, Claus M, Vandewalle V, Lenglez S, Essaghir A, Demoulin J-B, Havelange V. MiR-15a-5p Confers Chemoresistance in Acute Myeloid Leukemia by Inhibiting Autophagy Induced by Daunorubicin. International Journal of Molecular Sciences. 2021; 22(10):5153. https://doi.org/10.3390/ijms22105153
Chicago/Turabian StyleBollaert, Emeline, Melissa Claus, Virginie Vandewalle, Sandrine Lenglez, Ahmed Essaghir, Jean-Baptiste Demoulin, and Violaine Havelange. 2021. "MiR-15a-5p Confers Chemoresistance in Acute Myeloid Leukemia by Inhibiting Autophagy Induced by Daunorubicin" International Journal of Molecular Sciences 22, no. 10: 5153. https://doi.org/10.3390/ijms22105153
APA StyleBollaert, E., Claus, M., Vandewalle, V., Lenglez, S., Essaghir, A., Demoulin, J.-B., & Havelange, V. (2021). MiR-15a-5p Confers Chemoresistance in Acute Myeloid Leukemia by Inhibiting Autophagy Induced by Daunorubicin. International Journal of Molecular Sciences, 22(10), 5153. https://doi.org/10.3390/ijms22105153