
Overview of Cellular Immunotherapies within Transfusion Medicine for the Treatment of Malignant Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
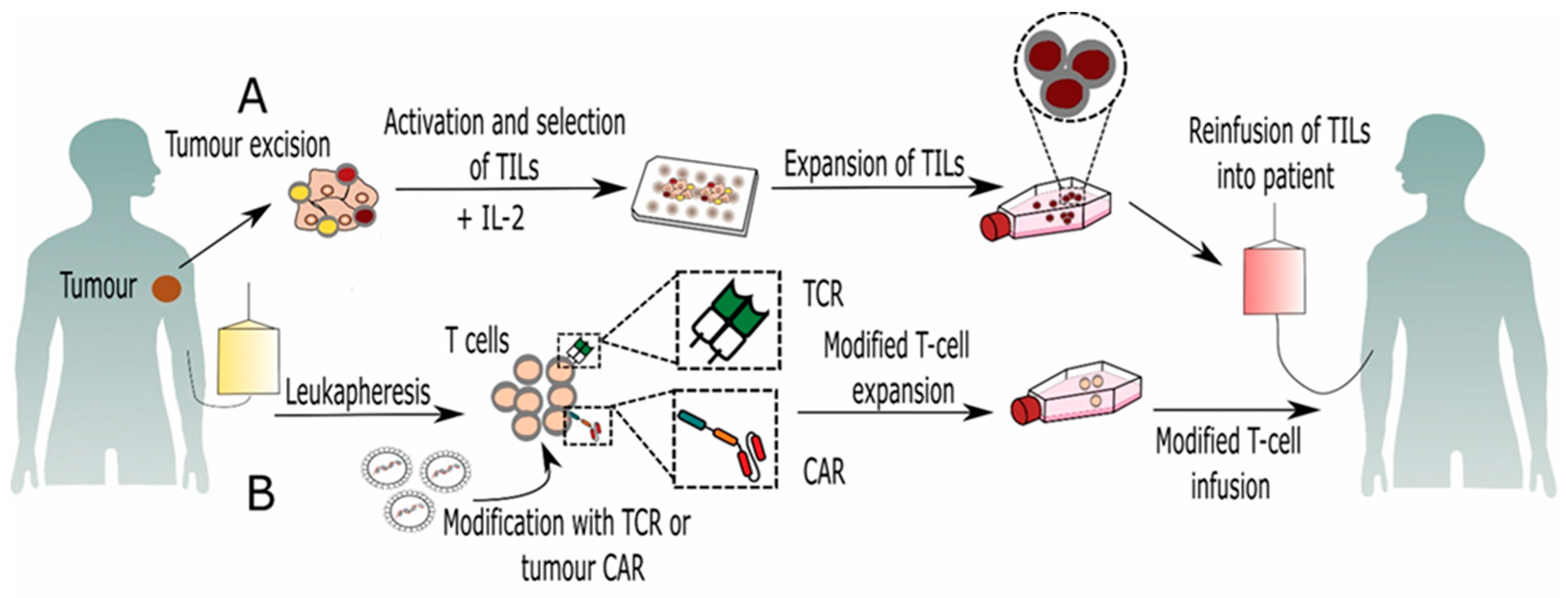
2. Tumor Infiltrating Lymphocytes (TILs)
3. Genetically Modified T Lymphocytes
3.1. CAR-T Cell-Based Immunotherapy
3.2. TCR-Based Immunotherapy
4. Other Adoptive T-Cell Therapies
4.1. γ/δT Cell-Based Immunotherapy
4.2. Cytotoxic T-Cell (CTL)-Based Immunotherapy
4.3. Donor Lymphocyte Infusion (DLI)-Based Immunotherapy
5. NK Cell-Based Immunotherapy
5.1. Adoptive Transfer of Unmodified NK Cells
5.2. Genetically Modified NK Cells
6. Cytokine-Induced Killer Cell (CIK)-Based Immunotherapy
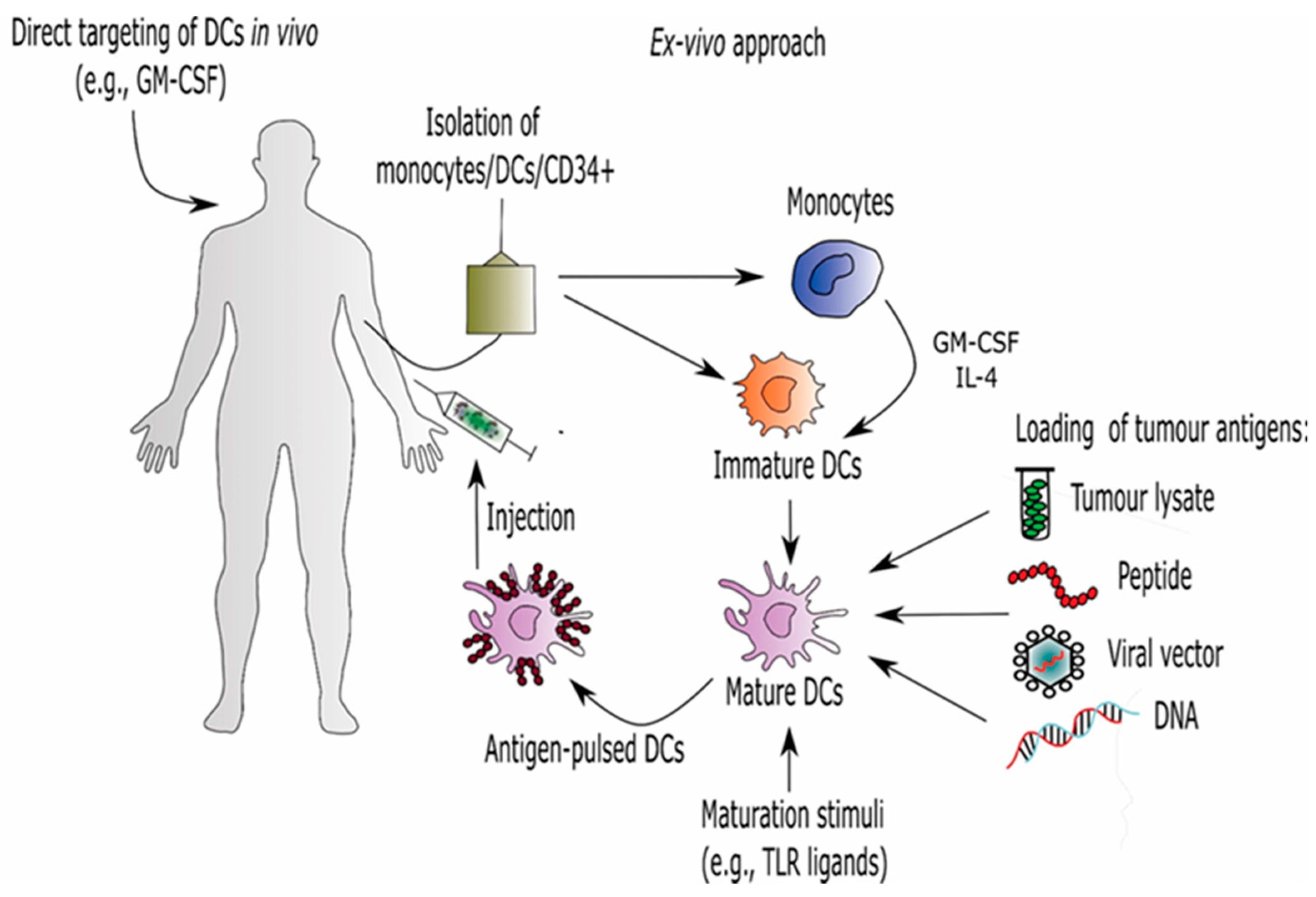
7. Dendritic Cell (DC)-Based Immunotherapy
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Franchini, M.; Capuzzo, E.; Turdo, R.; Glingani, C. Quality of transfusion products in blood banking. Semin. Thromb. Hemost. 2014, 40, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Seifried, E.; Mueller, M.M. The present and future of Transfusion Medicine. Blood Transfus. 2011, 9, 371–376. [Google Scholar] [PubMed]
- Marcucci, C.; Madjdpour, C.; Spahn, D.R. Allogeneic blood transfusions: Benefit, risks and clinical indications in countries with a low or high human development index. Br. Med. Bull. 2004, 70, 15–28. [Google Scholar] [CrossRef]
- Prigent, A.; Maillard, N.; Absi, L.; Aloui, C.; Cognasse, F.; Laradi, S.; Mariat, C.; Garraud, O. From Donor to Recipient: Current Questions Relating to Humoral Alloimmunization. Antibodies 2014, 3, 130–152. [Google Scholar] [CrossRef]
- Petricciani, J.; Hayakawa, T.; Stacey, G.; Trouvin, J.H.; Knezevic, I. Scientific considerations for the regulatory evaluation of cell therapy products. Biologicals 2017, 50, 20–26. [Google Scholar] [CrossRef]
- Koh, M.B.; Suck, G. Cell therapy: Promise fulfilled? Biologicals 2012, 40, 214–217. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, (Suppl 35), S185–S198. [Google Scholar] [CrossRef]
- Berd, D.; Maguire, H.C., Jr.; McCue, P.; Mastrangelo, M.J. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: Clinical and immunologic results in 64 patients. J. Clin. Oncol. 1990, 8, 1858–1867. [Google Scholar] [CrossRef]
- Lee, N.; Zakka, L.R.; Mihm, M.C., Jr.; Schatton, T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016, 48, 177–187. [Google Scholar] [CrossRef]
- Jiang, D.; Liu, Y.; Wang, H.; Wang, H.; Song, Q.; Sujie, A.; Huang, J.; Xu, Y.; Zeng, H.; Tan, L.; et al. Tumour infiltrating lymphocytes correlate with improved survival in patients with esophageal squamous cell carcinoma. Sci. Rep. 2017, 7, 44823. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef]
- Feng, W.; Li, Y.; Shen, L.; Cai, X.W.; Zhu, Z.F.; Chang, J.H.; Xiang, J.Q.; Zhang, Y.W.; Chen, H.Q.; Fu, X.L. Prognostic value of tumor-infiltrating lymphocytes for patients with completely resected stage IIIA(N2) non-small cell lung cancer. Oncotarget 2016, 7, 7227–7240. [Google Scholar] [CrossRef]
- Pennock, G.K.; Chow, L.Q. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014, 6, 1265–1278. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef]
- Mayor, P.; Starbuck, K.; Zsiros, E. Adoptive cell transfer using autologous tumor infiltrating lymphocytes in gynecologic malignancies. Gynecol. Oncol. 2018, 150, 361–369. [Google Scholar] [CrossRef]
- Zorn, E.; Mohseni, M.; Kim, H.; Porcheray, F.; Lynch, A.; Bellucci, R.; Canning, C.; Alyea, E.P.; Soiffer, R.J.; Ritz, J. Combined CD4+ donor lymphocyte infusion and low-dose recombinant IL-2 expand FOXP3+ regulatory T cells following allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2009, 15, 382–388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sim, G.C.; Martin-Orozco, N.; Jin, L.; Yang, Y.; Wu, S.; Washington, E.; Sanders, D.; Lacey, C.; Wang, Y.; Vence, L.; et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Investig. 2014, 124, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Holmich, L.R.; Hendel, H.W.; Met, O.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Model. Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Ye, B.; Stary, C.M.; Gao, Q.; Wang, Q.; Zeng, Z.; Jian, Z.; Gu, L.; Xiong, X. Genetically Modified T-Cell-Based Adoptive Immunotherapy in Hematological Malignancies. J. Immunol. Res. 2017, 2017, 5210459. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Lee, D.A.; Denman, C.J.; Rondon, G.; Woodworth, G.; Chen, J.; Fisher, T.; Kaur, I.; Fernandez-Vina, M.; Cao, K.; Ciurea, S.; et al. Haploidentical Natural Killer Cells Infused before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biol. Blood Marrow Transplant. 2016, 22, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; De Leon, G.; Boczkowski, D.; Schmittling, R.; Xie, W.; Staats, J.; Liu, R.; Johnson, L.A.; Weinhold, K.; Archer, G.E.; et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin. Cancer Res. 2014, 20, 2684–2694. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Treatment of Cancer Brain T, Radiotherapy G, National Cancer Institute of Canada Clinical Trials G. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Morris, E.C.; Stauss, H.J. Optimizing T-cell receptor gene therapy for hematologic malignancies. Blood 2016, 127, 3305–3311. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, X.; Wang, L.; Gao, X.; Xiong, Y.; Liu, L.; Wei, F.; Yang, L.; Ren, X. T-cell receptor gene therapy targeting melanoma-associated antigen-A4 by silencing of endogenous TCR inhibits tumor growth in mice and human. Cell Death Dis. 2019, 10, 475. [Google Scholar] [CrossRef]
- Gogoi, D.; Chiplunkar, S.V. Targeting gamma delta T cells for cancer immunotherapy: Bench to bedside. Indian J. Med. Res. 2013, 138, 755–761. [Google Scholar]
- Zhao, Y.; Niu, C.; Cui, J. Gamma-delta (γδ) T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef]
- Wilhelm, M.; Kunzmann, V.; Eckstein, S.; Reimer, P.; Weissinger, F.; Ruediger, T.; Tony, H.P. Γδ T cells for immune therapy of patients with lymphoid malignancies. Blood 2003, 102, 200–206. [Google Scholar] [CrossRef]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Adams, J.; Harkins, L.; Kelly, D.; You, Z.; Lamb, L.S., Jr. In vivo expansion and activation of γδ T cells as immunotherapy for refractory neuroblastoma: A phase 1 study. Medicine 2016, 95, e4909. [Google Scholar] [CrossRef]
- Nicol, A.J.; Tokuyama, H.; Mattarollo, S.R.; Hagi, T.; Suzuki, K.; Yokokawa, K.; Nieda, M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br. J. Cancer 2011, 105, 778–786. [Google Scholar] [CrossRef]
- Wilhelm, M.; Smetak, M.; Schaefer-Eckart, K.; Kimmel, B.; Birkmann, J.; Einsele, H.; Kunzmann, V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014, 12, 45. [Google Scholar] [CrossRef]
- Wada, I.; Matsushita, H.; Noji, S.; Mori, K.; Yamashita, H.; Nomura, S.; Shimizu, N.; Seto, Y.; Kakimi, K. Intraperitoneal injection of in vitro expanded Vgamma9Vdelta2 T cells together with zoledronate for the treatment of malignant ascites due to gastric cancer. Cancer Med. 2014, 3, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Meidenbauer, N.; Vogl, S.; Laumer, M.; Berger, J.; Andreesen, R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5060–5069. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Zhao, Q.; Chen, G.; Chen, J.; Yan, X.; Zhou, Y.H.; Huang, Z. Soluble CD40 ligand-activated human peripheral B cells as surrogated antigen presenting cells: A preliminary approach for anti-HBV immunotherapy. Virol. J. 2010, 7, 370. [Google Scholar] [CrossRef] [PubMed]
- Neal, L.R.; Bailey, S.R.; Wyatt, M.M.; Bowers, J.S.; Majchrzak, K.; Nelson, M.H.; Haupt, C.; Paulos, C.M.; Varela, J.C. The Basics of Artificial Antigen Presenting Cells in T Cell-Based Cancer Immunotherapies. J. Immunol. Res. Ther. 2017, 2, 68–79. [Google Scholar] [PubMed]
- Suhoski, M.M.; Golovina, T.N.; Aqui, N.A.; Tai, V.C.; Varela-Rohena, A.; Milone, M.C.; Carroll, R.G.; Riley, J.L.; June, C.H. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol. Ther. 2007, 15, 981–988. [Google Scholar] [CrossRef]
- Butler, M.O.; Lee, J.S.; Ansen, S.; Neuberg, D.; Hodi, F.S.; Murray, A.P.; Drury, L.; Berezovskaya, A.; Mulligan, R.C.; Nadler, L.M.; et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin. Cancer Res. 2007, 13, 1857–1867. [Google Scholar] [CrossRef]
- Oelke, M.; Maus, M.V.; Didiano, D.; June, C.H.; Mackensen, A.; Schneck, J.P. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat. Med. 2003, 9, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Bieler, J.G.; Schutz, C.; Varela, J.C.; Douglass, J.; Skora, A.; Chiu, Y.L.; Oelke, M.; Kinzler, K.; Zhou, S.; et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano 2015, 9, 6861–6871. [Google Scholar] [CrossRef]
- Hickey, J.W.; Vicente, F.P.; Howard, G.P.; Mao, H.Q.; Schneck, J.P. Biologically Inspired Design of Nanoparticle Artificial Antigen-Presenting Cells for Immunomodulation. Nano Lett. 2017, 17, 7045–7054. [Google Scholar] [CrossRef]
- Leung, W.; Soh, T.G.; Linn, Y.C.; Low, J.G.; Loh, J.; Chan, M.; Chng, W.J.; Koh, L.P.; Poon, M.L.; Ng, K.P.; et al. Rapid production of clinical-grade SARS-CoV-2 specific T cells. Adv. Cell Gene Ther. 2020, e101. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J.; Mittermuller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.H., Jr.; Shpilberg, O.; Drobyski, W.R.; Porter, D.L.; Giralt, S.; Champlin, R.; Goodman, S.A.; Wolff, S.N.; Hu, W.; Verfaillie, C.; et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J. Clin. Oncol. 1997, 15, 433–444. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Nagler, A.; Bornhauser, M.; Finke, J.; Fassas, A.; Volin, L.; Gurman, G.; Maertens, J.; Bordigoni, P.; et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: A retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J. Clin. Oncol. 2007, 25, 4938–4945. [Google Scholar] [PubMed]
- Nikiforow, S.; Kim, H.T.; Daley, H.; Reynolds, C.; Jones, K.T.; Armand, P.; Ho, V.T.; Alyea, E.P., 3rd; Cutler, C.S.; Ritz, J.; et al. A phase I study of CD25/regulatory T-cell-depleted donor lymphocyte infusion for relapse after allogeneic stem cell transplantation. Haematologica 2016, 101, 1251–1259. [Google Scholar] [CrossRef]
- Durer, S.; Durer, C.; Shafqat, M.; Comba, I.Y.; Malik, S.; Faridi, W.; Aslam, S.; Ijaz, A.; Tariq, M.J.; Fraz, M.A.; et al. Concomitant use of blinatumomab and donor lymphocyte infusion for mixed-phenotype acute leukemia: A case report with literature review. Immunotherapy 2019, 11, 373–378. [Google Scholar] [CrossRef]
- Park, H.B.; Lee, J.E.; Oh, Y.M.; Lee, S.J.; Eom, H.S.; Choi, K. CTLA4-CD28 chimera gene modification of T cells enhances the therapeutic efficacy of donor lymphocyte infusion for hematological malignancy. Exp. Mol. Med. 2017, 49, e360. [Google Scholar] [CrossRef][Green Version]
- Hashimoto, H.; Kitano, S.; Ueda, R.; Ito, A.; Tada, K.; Fuji, S.; Yamashita, T.; Tomura, D.; Nukaya, I.; Mineno, J.; et al. Infusion of donor lymphocytes expressing the herpes simplex virus thymidine kinase suicide gene for recurrent hematologic malignancies after allogeneic hematopoietic stem cell transplantation. Int. J. Hematol. 2015, 102, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsis-Mortari, A.; Keever-Taylor, C.A.; Zhang, M.J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow Transplant. 2003, 32, 177–186. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.T.; Carlsten, M.; Sohlberg, E.; Liu, L.L.; Clancy, T.; Karimi, M.; Cooley, S.; Miller, J.S.; Klimkowska, M.; Schaffer, M.; et al. Complete Remission with Reduction of High-Risk Clones following Haploidentical NK-Cell Therapy against MDS and AML. Clin. Cancer Res. 2018, 24, 1834–1844. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Ishikawa, T.; Okayama, T.; Sakamoto, N.; Ideno, M.; Oka, K.; Enoki, T.; Mineno, J.; Yoshida, N.; Katada, K.; Kamada, K.; et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 2018, 142, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Cooley, S.; He, F.; Bachanova, V.; Vercellotti, G.M.; DeFor, T.E.; Curtsinger, J.M.; Robertson, P.; Grzywacz, B.; Conlon, K.C.; Waldmann, T.A.; et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019, 3, 1970–1980. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Michel, G.; Galambrun, C.; Sirvent, A.; Pochon, C.; Bruno, B.; Jubert, C.; Loundou, A.; Yakoub-Agha, I.; Milpied, N.; Lutz, P.; et al. Single- vs double-unit cord blood transplantation for children and young adults with acute leukemia or myelodysplastic syndrome. Blood 2016, 127, 3450–3457. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; den Hollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M., 2nd; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jaw, J.J.; Stutzman, N.C.; Zou, Z.; Sun, P.D. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012, 91, 299–309. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Le Luduec, J.B.; Forlenza, C.; Jakubowski, A.A.; Perales, M.A.; Young, J.W.; Hsu, K.C. Phase II Study of Haploidentical Natural Killer Cell Infusion for Treatment of Relapsed or Persistent Myeloid Malignancies Following Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 705–709. [Google Scholar] [CrossRef]
- Muller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Foltz, J.A.; Hess, B.T.; Bachanova, V.; Bartlett, N.L.; Berrien-Elliott, M.M.; McClain, E.; Becker-Hapak, M.K.; Foster, M.; Schappe, T.; Kahl, B.; et al. Phase 1 trial of N-803, an IL-15 receptor agonist, with rituximab in patients with indolent non-Hodgkin lymphoma. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Wolf, I.G.; Finke, S.; Trojaneck, B.; Denkena, A.; Lefterova, P.; Schwella, N.; Heuft, H.G.; Prange, G.; Korte, M.; Takeya, M.; et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br. J. Cancer 1999, 81, 1009–1016. [Google Scholar] [CrossRef]
- Li, D.P.; Li, W.; Feng, J.; Chen, K.; Tao, M. Adjuvant chemotherapy with sequential cytokine-induced killer (CIK) cells in stage IB non-small cell lung cancer. Oncol. Res. 2015, 22, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Wei, F.; An, X.; Zhang, N.; Cao, S.; Ren, B.; Zhang, X.; Ren, X. Efficiency of Cytokine-Induced Killer Cells in Combination with Chemotherapy for Triple-Negative Breast Cancer. J. Breast Cancer 2018, 21, 150–157. [Google Scholar] [CrossRef]
- Lin, T.; Song, C.; Chuo, D.Y.; Zhang, H.; Zhao, J. Clinical effects of autologous dendritic cells combined with cytokine-induced killer cells followed by chemotherapy in treating patients with advanced colorectal cancer: A prospective study. Tumour Biol. 2016, 37, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Ma, W.; Lu, H.; Yuan, L.; An, L.; Wang, X.; Cheng, G.; Zuo, S. Modification of cytokine-induced killer cells with chimeric antigen receptors (CARs) enhances antitumor immunity to epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol. Immunother. 2015, 64, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Merker, M.; Pfirrmann, V.; Oelsner, S.; Fulda, S.; Klingebiel, T.; Wels, W.S.; Bader, P.; Rettinger, E. Generation and characterization of ErbB2-CAR-engineered cytokine-induced killer cells for the treatment of high-risk soft tissue sarcoma in children. Oncotarget 2017, 8, 66137–66153. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J. Exp. Med. 1974, 139, 380–397. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef]
- Zhou, L.J.; Tedder, T.F. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2588–2592. [Google Scholar] [CrossRef]
- Ratta, M.; Rondelli, D.; Fortuna, A.; Curti, A.; Fogli, M.; Fagnoni, F.; Martinelli, G.; Terragna, C.; Tura, S.; Lemoli, R.M. Generation and functional characterization of human dendritic cells derived from CD34 cells mobilized into peripheral blood: Comparison with bone marrow CD34+ cells. Br. J. Haematol. 1998, 101, 756–765. [Google Scholar] [CrossRef]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017, 7, 2091–2102. [Google Scholar] [PubMed]
- Freitas-Silva, R.; Brelaz-de-Castro, M.C.; Pereira, V.R. Dendritic cell-based approaches in the fight against diseases. Front. Immunol. 2014, 5, 78. [Google Scholar] [CrossRef]
- Mukherji, B.; Chakraborty, N.G.; Yamasaki, S.; Okino, T.; Yamase, H.; Sporn, J.R.; Kurtzman, S.K.; Ergin, M.T.; Ozols, J.; Meehan, J.; et al. Induction of antigen-specific cytolytic T cells in situ in human melanoma by immunization with synthetic peptide-pulsed autologous antigen presenting cells. Proc. Natl. Acad. Sci. USA 1995, 92, 8078–8082. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.F.; Punt, C.J.; Lesterhuis, W.J.; Sutmuller, R.P.; Brouwer, H.M.; Scharenborg, N.M.; Klasen, I.S.; Hilbrands, L.B.; Figdor, C.G.; de Vries, I.J.; et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: A phase I/II study in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 5067–5078. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Furuse, M.; Nonoguchi, N.; Omura, N.; Shirahata, M.; Iwasaki, K.; Inui, T.; Kuroiwa, T.; Kuwabara, H.; Miyatake, S.-I. Immunotherapy of Nivolumab with Dendritic Cell Vaccination Is Effective against Intractable Recurrent Primary Central Nervous System Lymphoma: A Case Report. Neurol. Med. Chir. 2017, 57, 191–197. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma-A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [PubMed]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Xi, H.B.; Wang, G.X.; Fu, B.; Liu, W.P.; Li, Y. Survivin and PSMA Loaded Dendritic Cell Vaccine for the Treatment of Prostate Cancer. Biol. Pharm. Bull. 2015, 38, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tešić, N.; Poženel, P.; Švajger, U. Overview of Cellular Immunotherapies within Transfusion Medicine for the Treatment of Malignant Diseases. Int. J. Mol. Sci. 2021, 22, 5120. https://doi.org/10.3390/ijms22105120
Tešić N, Poženel P, Švajger U. Overview of Cellular Immunotherapies within Transfusion Medicine for the Treatment of Malignant Diseases. International Journal of Molecular Sciences. 2021; 22(10):5120. https://doi.org/10.3390/ijms22105120
Chicago/Turabian StyleTešić, Nataša, Primož Poženel, and Urban Švajger. 2021. "Overview of Cellular Immunotherapies within Transfusion Medicine for the Treatment of Malignant Diseases" International Journal of Molecular Sciences 22, no. 10: 5120. https://doi.org/10.3390/ijms22105120
APA StyleTešić, N., Poženel, P., & Švajger, U. (2021). Overview of Cellular Immunotherapies within Transfusion Medicine for the Treatment of Malignant Diseases. International Journal of Molecular Sciences, 22(10), 5120. https://doi.org/10.3390/ijms22105120