
Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing?


Abstract
1. Introduction
2. Treatment of Melanoma
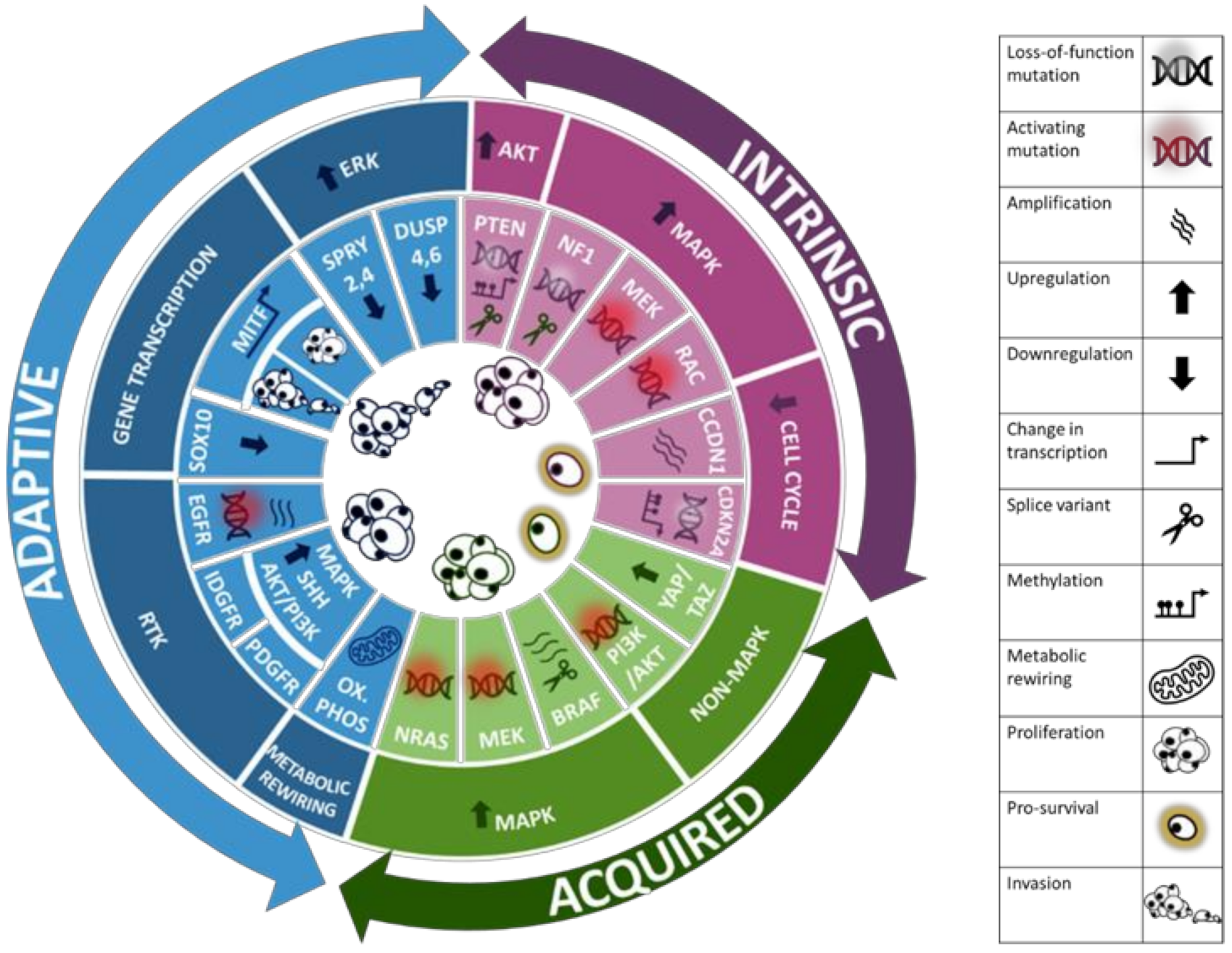
3. Resistance Mechanisms to Targeted Therapy in Melanoma
3.1. Intrinsic Resistance
3.1.1. PI3K/AKT Pathway Activation
3.1.2. MAPK Pathway Activation
3.1.3. Disruptions to the Cell Cycle
3.2. Adaptive Resistance
3.2.1. ERK Signalling Reactivation
3.2.2. Alteration of Gene Transcription
3.2.3. Alteration of RTK Signalling
3.2.4. Metabolic Rewiring
3.3. Acquired Resistance
3.3.1. Secondary Mutations in the MAPK Pathway
3.3.2. Activation of Non-MAPK Proliferative Signalling
4. The Tumour Suppressor, RASSF1A
RASSF1A and Melanoma
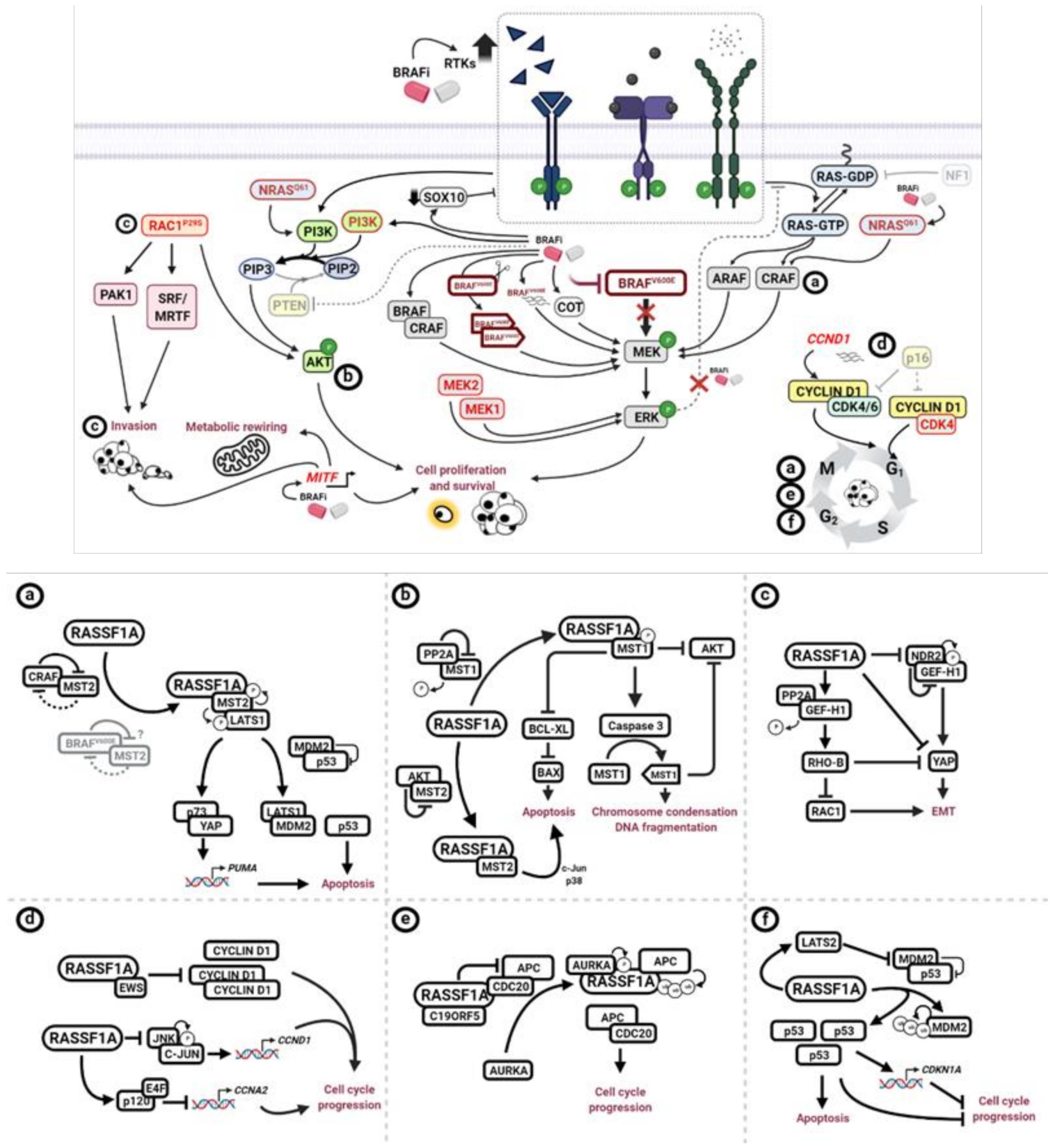
5. Potential Impact of RASSF1A Loss on BRAFi Targeted Therapy Resistance
5.1. MAPK Pathway
5.2. PTEN Loss and PI3K/AKT Hyperactivation
5.3. RAC1 Hyperactivation
5.4. CCND1 Amplification and CDNK2A Loss
6. Restoration of RASSF1A Expression by Demethylating Agents
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, R.J.; Rigel, D.S.; Kopf, A.W. Early Detection of Malignant Melanoma: The Role of Physician Examination and Self-Examination of the Skin. Ca A Cancer J. Clin. 1985, 35, 130–151. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.F.; Azizi, E.; Bianchi-Scarra, G.; Bishop, D.T.; Bressac-De Paillerets, B.; et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.J.; Farber, M.J.; Warycha, M.A.; Papathasis, N.; Miller, M.K.; Heilman, E.R. The “dysplastic” nevus. Clin. Dermatol. 2009, 27, 103–115. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Tucker, M.A. Melanoma Epidemiology. Hematol. Clin. N. Am. 2009, 23, 383–395. [Google Scholar] [CrossRef]
- Psaty, E.L.; Scope, A.; Halpern, A.C.; Marghoob, A.A. Defining the patient at high risk for melanoma. Int. J. Dermatol. 2010, 49, 362–376. [Google Scholar] [CrossRef]
- Riker, A.I.; Zea, N.; Trinh, T. The epidemiology, prevention, and detection of melanoma. Ochsner J. 2010, 10, 56–65. [Google Scholar]
- Scolyer, R.A.; Long, G.V.; Thompson, J.F. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Mol. Oncol. 2011, 5, 124–136. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Parker, J.F.; Florell, S.R.; Alexander, A.; DiSario, J.A.; Shami, P.J.; Leachman, S.A. Pancreatic carcinoma surveillance in patients with familial melanoma. Arch. Dermatol. 2003, 139, 1019–1025. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S.M. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug resistance of BRAF-mutant melanoma: Review of up-to-date mechanisms of action and promising targeted agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Mitogen-Activated Protein Kinase Activation in UV-Induced Signal Transduction. Sci. Signal. 2003, 2003, re2. [Google Scholar] [CrossRef]
- Houghton, A.N.; Polsky, D. Focus on melanoma. Cancer Cell 2002, 2, 275–278. [Google Scholar] [CrossRef]
- Lee, C.; Collichio, F.; Ollila, D.; Moschos, S. Historical review of melanoma treatment and outcomes. Clin. Dermatol. 2013, 31, 141–147. [Google Scholar] [CrossRef]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A., Jr.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef]
- Damsky, W.E.; Rosenbaum, L.E.; Bosenberg, M. Decoding melanoma metastasis. Cancers 2011, 3, 126–163. [Google Scholar] [CrossRef]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Maverakis, E.; Cornelius, L.; Bowen, G.; Phan, T.; Patel, F.; Fitzmaurice, S.; He, Y.; Burrall, B.; Duong, C.; Kloxin, A.; et al. Metastatic Melanoma—A Review of Current and Future Treatment Options. Acta Derm. Venereol. 2015, 95, 516–524. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAF V600–mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Latimer, N.R.; Abrams, K.R.; Amonkar, M.M.; Stapelkamp, C.; Swann, R.S. Adjusting for the Confounding Effects of Treatment Switching—The BREAK-3 Trial: Dabrafenib Versus Dacarbazine. Oncologist 2015, 20, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. Oncotargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; Preclinical Characterization, Increased Efficacy when Combined with Trametinib, while BRAF/MEK Tool Combination Reduced Skin Lesions. PLoS ONE 2013, 8, e67583. [Google Scholar] [CrossRef]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant—A mini review. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Tagliaferri, L.; Lancellotta, V.; Fionda, B.; Mangoni, M.; Casa, C.; Di Stefani, A.; Pagliara, M.M.; D’Aviero, A.; Schinzari, G.; Chiesa, S.; et al. Immunotherapy and radiotherapy in melanoma: A multidisciplinary comprehensive review. Hum. Vaccines Immunother. 2021, 1–8. [Google Scholar] [CrossRef]
- Hecht, M.; Meier, F.; Zimmer, L.; Polat, B.; Loquai, C.; Weishaupt, C.; Forschner, A.; Gutzmer, R.; Utikal, J.S.; Goldinger, S.M.; et al. Clinical outcome of concomitant vs interrupted BRAF inhibitor therapy during radiotherapy in melanoma patients. Br. J. Cancer 2018, 118, 785–792. [Google Scholar] [CrossRef]
- Chowdhary, M.; Patel, K.R.; Danish, H.H.; Lawson, D.H.; Khan, M.K. BRAF inhibitors and radiotherapy for melanoma brain metastases: Potential advantages and disadvantages of combination therapy. Onco Targets Ther. 2016, 9, 7149–7159. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Lioni, M.; Dalla Palma, M.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Piglowski, W.; Rajczykowski, M.; Pfeifer, A.; Student, S.; Fiszer-Kierzkowska, A. Genetic Profiling of Advanced Melanoma: Candidate Mutations for Predicting Sensitivity and Resistance to Targeted Therapy. Target. Oncol. 2020, 15, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Catalanotti, F.; Cheng, D.T.; Shoushtari, A.N.; Johnson, D.B.; Panageas, K.S.; Momtaz, P.; Higham, C.; Won, H.H.; Harding, J.J.; Merghoub, T.; et al. PTEN Loss-of-Function Alterations Are Associated with Intrinsic Resistance to BRAF Inhibitors in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Montagut, C.; Sharma, S.V.; Shioda, T.; McDermott, U.; Ulman, M.; Ulkus, L.E.; Dias-Santagata, D.; Stubbs, H.; Lee, D.Y.; Singh, A.; et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008, 68, 4853–4861. [Google Scholar] [CrossRef]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Carlino, M.S.; Fung, C.; Shahheydari, H.; Todd, J.R.; Boyd, S.C.; Irvine, M.; Nagrial, A.M.; Scolyer, R.A.; Kefford, R.F.; Long, G.V.; et al. Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin. Cancer Res. 2015, 21, 98–105. [Google Scholar] [CrossRef]
- Py, C.; Christinat, Y.; Kreutzfeldt, M.; McKee, T.A.; Dietrich, P.-Y.; Tsantoulis, P. Response of NF1-Mutated Melanoma to an MEK Inhibitor. JCO Precis. Oncol. 2018, 1–11. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., II; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef]
- Halaban, R. RAC1 and melanoma. Clin. Ther. 2015, 37, 682–685. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1(P29S) Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e69. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of acquired BRAF inhibitor resistance in melanoma: A systematic review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical Interaction with Yes-associated Protein Enhances p73 Transcriptional Activity*. J. Biol. Chem. 2001. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted therapy in melanoma and mechanisms of resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, W. A review of the molecular pathways involved in resistance to BRAF inhibitors in patients with advanced-stage melanoma. Med. Sci. Monit. 2020, 26, e920957-1–e920957-13. [Google Scholar] [CrossRef]
- Swayden, M.; Chhouri, H.; Anouar, Y.; Grumolato, L. Tolerant/Persister Cancer Cells and the Path to Resistance to Targeted Therapy. Cells 2020, 9, 2601. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef]
- Cheli, Y.; Ohanna, M.; Ballotti, R.; Bertolotto, C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment. Cell Melanoma Res. 2010, 23, 27–40. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef]
- Möller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Ren, Y.; He, W.; Liu, H.; Zhi, Z.; Zhu, X.; Yang, T.; Rong, Y.; Ma, B.; Purwin, T.J.; et al. ERK-mediated phosphorylation regulates SOX10 sumoylation and targets expression in mutant BRAF melanoma. Nat. Commun. 2018, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Wang, Y.; Wang, X.; Flaherty, K.T.; Yu, L.; Pepin, D.; Scognamiglio, G.; Pepe, S.; Kirkwood, J.M.; Cooper, Z.A.; et al. PDGFRa up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget 2014, 5, 1926–1941. [Google Scholar] [CrossRef]
- Villanueva, J.; Infante, J.R.; Krepler, C.; Reyes-Uribe, P.; Samanta, M.; Chen, H.Y.; Li, B.; Swoboda, R.K.; Wilson, M.; Vultur, A.; et al. Concurrent MEK2 Mutation and BRAF Amplification Confer Resistance to BRAF and MEK Inhibitors in Melanoma. Cell Rep. 2013, 4, 1090–1099. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Romano, E.; Pradervand, S.; Paillusson, A.; Weber, J.; Harshman, K.; Muehlethaler, K.; Speiser, D.; Peters, S.; Rimoldi, D.; Michielin, O. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin. Cancer Res. 2013, 19, 5749–5757. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Ahmed, T.A.; Tucker, M.R.; Ung, P.M.U.; Xiao, M.; Karoulia, Z.; Amabile, A.; Wu, X.; Aaronson, S.A.; Ang, C.; et al. Exploiting allosteric properties of RAF and MEK inhibitors to target therapy-resistant tumors driven by oncogenic BRAF signaling. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef]
- Arora, R.; Di Michele, M.; Stes, E.; Vandermarliere, E.; Martens, L.; Gevaert, K.; Van Heerde, E.; Linders, J.T.M.; Brehmer, D.; Jacoby, E.; et al. Structural investigation of B-Raf paradox breaker and inducer inhibitors. J. Med. Chem. 2015, 58, 1818–1831. [Google Scholar] [CrossRef]
- Callus, B.A.; Finch-Edmondson, M.L.; Fletcher, S.; Wilton, S.D. YAPping about and not forgetting TAZ. FEBS Lett. 2019, 593, 253–276. [Google Scholar] [CrossRef]
- Nakatani, K.; Maehama, T.; Nishio, M.; Goto, H.; Kato, W.; Omori, H.; Miyachi, Y.; Togashi, H.; Shimono, Y.; Suzuki, A. Targeting the Hippo signalling pathway for cancer treatment. J. Biochem. 2017, 161, 237–244. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, L.; McKenna, S.; Kolch, W.; Matallanas, D. RASSF1A Tumour Suppressor: Target the Network for Effective Cancer Therapy. Cancers 2020, 12, 229. [Google Scholar] [CrossRef]
- Bradley, L.; Koennig, D.; Tognoli, M.L.; van der Vaart, J.; O’Neill, E. RASSF Family. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 4501–4523. [Google Scholar]
- Dubois, F.; Bergot, E.; Levallet, G. Cancer and RASSF1A/RASSF1C, the Two Faces of Janus. Trends Cancer 2019, 5, 662–665. [Google Scholar] [CrossRef]
- Dubois, F.; Bergot, E.; Zalcman, G.; Levallet, G. RASSF1A, puppeteer of cellular homeostasis, fights tumorigenesis, and metastasis—An updated review. Cell Death Dis. 2019, 10, 928. [Google Scholar] [CrossRef]
- Pefani, D.E.; O’Neill, E. Safeguarding genome stability: RASSF1A tumor suppressor regulates BRCA2 at stalled forks. Cell Cycle 2015, 14, 1624–1630. [Google Scholar] [CrossRef]
- Dallol, A.; Agathanggelou, A.; Fenton, S.L.; Ahmed-Choudhury, J.; Hesson, L.; Vos, M.D.; Clark, G.J.; Downward, J.; Maher, E.R.; Latif, F. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res. 2004, 64, 4112–4116. [Google Scholar] [CrossRef]
- Donninger, H.; Clark, J.A.; Monaghan, M.K.; Schmidt, M.L.; Vos, M.; Clark, G.J. Cell cycle restriction is more important than apoptosis induction for RASSF1A protein tumor suppression. J. Biol. Chem. 2014, 289, 31287–31295. [Google Scholar] [CrossRef]
- Chow, C.; Wong, N.; Pagano, M.; Lun, S.W.; Nakayama, K.I.; Nakayama, K.; Lo, K.W. Regulation of APC/CCdc20 activity by RASSF1A-APC/CCdc20 circuitry. Oncogene 2012, 31, 1975–1987. [Google Scholar] [CrossRef]
- Song, M.S.; Song, S.J.; Ayad, N.G.; Chang, J.S.; Lee, J.H.; Hong, H.K.; Lee, H.; Choi, N.; Kim, J.; Kim, H.; et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Whitehurst, A.W.; Ram, R.; Shivakumar, L.; Gao, B.; Minna, J.D.; White, M.A. The RASSF1A tumor suppressor restrains anaphase-promoting complex/cyclosome activity during the G1/S phase transition to promote cell cycle progression in human epithelial cells. Mol. Cell. Biol. 2008, 28, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Choudhury, J.; Agathanggelou, A.; Fenton, S.L.; Ricketts, C.; Clark, G.J.; Maher, E.R.; Latif, F. Transcriptional regulation of cyclin A2 by RASSF1A through the enhanced binding of p120E4F to the cyclin A2 promoter. Cancer Res. 2005, 65, 2690–2697. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fenton, S.L.; Dallol, A.; Agathanggelou, A.; Hesson, L.; Ahmed-Choudhury, J.; Baksh, S.; Sardet, C.; Dammann, R.; Minna, J.D.; Downward, J.; et al. Identification of the E1A-regulated transcription factor p120 E4F as an interacting partner of the RASSF1A candidate tumor suppressor gene. Cancer Res. 2004, 64, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Lee, M.G.; Moon, J.R.; Lee, C.K.; Chi, S.G.; Kim, H.J. Ras association domain family 1 isoform A suppresses colonic tumor cell growth through p21(WAF1) activation in a p53-dependent manner. J. Gastroenterol. Hepatol. 2019, 34, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, L.; Minna, J.; Sakamaki, T.; Pestell, R.; White, M.A. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol. Cell. Biol. 2002, 22, 4309–4318. [Google Scholar] [CrossRef]
- Thaler, S.; Hahnel, P.S.; Schad, A.; Dammann, R.; Schuler, M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009, 69, 1748–1757. [Google Scholar] [CrossRef]
- Whang, Y.M.; Kim, Y.H.; Kim, J.S.; Yoo, Y.D. RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res. 2005, 65, 3682–3690. [Google Scholar] [CrossRef]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef]
- Foley, C.J.; Freedman, H.; Choo, S.L.; Onyskiw, C.; Fu, N.Y.; Yu, V.C.; Tuszynski, J.; Pratt, J.C.; Baksh, S. Dynamics of RASSF1A/MOAP-1 association with death receptors. Mol. Cell. Biol. 2008, 28, 4520–4535. [Google Scholar] [CrossRef]
- Tan, K.O.; Tan, K.M.; Chan, S.L.; Yee, K.S.; Bevort, M.; Ang, K.C.; Yu, V.C. MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with Bax through its Bcl-2 homology domains. J. Biol. Chem. 2001, 276, 2802–2807. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 2011, 44, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazieres, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. RASSF1A Suppresses the Invasion and Metastatic Potential of Human Non-Small Cell Lung Cancer Cells by Inhibiting YAP Activation through the GEF-H1/RhoB Pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.J.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef]
- Lee, M.G.; Jeong, S.I.; Ko, K.P.; Park, S.K.; Ryu, B.K.; Kim, I.Y.; Kim, J.K.; Chi, S.G. RASSF1A Directly Antagonizes RhoA Activity through the Assembly of a Smurf1-Mediated Destruction Complex to Suppress Tumorigenesis. Cancer Res. 2016, 76, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Cooper, W.N.; Latif, F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005, 65, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Grawenda, A.M.; O’Neill, E. Clinical utility of RASSF1A methylation in human malignancies. Br. J. Cancer 2015, 113, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Malpeli, G.; Innamorati, G.; Decimo, I.; Bencivenga, M.; Nwabo Kamdje, A.H.; Perris, R.; Bassi, C. Methylation Dynamics of RASSF1A and Its Impact on Cancer. Cancers 2019, 11, 959. [Google Scholar] [CrossRef]
- Dallol, A.; Agathanggelou, A.; Tommasi, S.; Pfeifer, G.P.; Maher, E.R.; Latif, F. Involvement of the RASSF1A tumor suppressor gene in controlling cell migration. Cancer Res. 2005, 65, 7653–7659. [Google Scholar] [CrossRef]
- Guo, Y.; Long, J.; Lei, S. Promoter methylation as biomarkers for diagnosis of melanoma: A systematic review and meta-analysis. J. Cell. Physiol. 2019, 234, 7356–7367. [Google Scholar] [CrossRef]
- Salvianti, F.; Orlando, C.; Massi, D.; De Giorgi, V.; Grazzini, M.; Pazzagli, M.; Pinzani, P. Tumor-Related Methylated Cell-Free DNA and Circulating Tumor Cells in Melanoma. Front. Mol. Biosci. 2015, 2, 76. [Google Scholar] [CrossRef]
- Xu, G.; Zhou, X.; Xing, J.; Xiao, Y.; Jin, B.; Sun, L.; Yang, H.; Du, S.; Xu, H.; Mao, Y. Identification of RASSF1A promoter hypermethylation as a biomarker for hepatocellular carcinoma. Cancer Cell Int. 2020, 20, 547. [Google Scholar] [CrossRef]
- Ye, M.; Huang, T.; Ni, C.; Yang, P.; Chen, S. Diagnostic Capacity of RASSF1A Promoter Methylation as a Biomarker in Tissue, Brushing, and Blood Samples of Nasopharyngeal Carcinoma. EBioMedicine 2017, 18, 32–40. [Google Scholar] [CrossRef]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef]
- Spugnardi, M.; Tommasi, S.; Dammann, R.; Pfeifer, G.P.; Hoon, D.S. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003, 63, 1639–1643. [Google Scholar]
- Reifenberger, J.; Knobbe, C.B.; Sterzinger, A.A.; Blaschke, B.; Schulte, K.W.; Ruzicka, T.; Reifenberger, G. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int J. Cancer 2004, 109, 377–384. [Google Scholar] [CrossRef]
- Rother, J.; Jones, D. Molecular markers of tumor progression in melanoma. Curr. Genom. 2009, 10, 231–239. [Google Scholar] [CrossRef][Green Version]
- Yi, M.; Yang, J.; Chen, X.; Li, J.; Li, X.; Wang, L.; Tan, Y.; Xiong, W.; Zhou, M.; McCarthy, J.B.; et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J. Cell. Physiol. 2011, 226, 2360–2369. [Google Scholar] [CrossRef]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef]
- Savoia, P.; Zavattaro, E.; Cremona, O. Clinical Implications of Acquired BRAF Inhibitors Resistance in Melanoma. Int. J. Mol. Sci. 2020, 21, 9730. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef]
- O’Neill, E.; Rushworth, L.; Baccarini, M.; Kolch, W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004, 306, 2267–2270. [Google Scholar] [CrossRef]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef]
- Romano, D.; Matallanas, D.; Weitsman, G.; Preisinger, C.; Ng, T.; Kolch, W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010, 70, 1195–1203. [Google Scholar] [CrossRef]
- Romano, D.; Maccario, H.; Doherty, C.; Quinn, N.P.; Kolch, W.; Matallanas, D. The Differential Effects of Wild-Type and Mutated K-Ras on MST2 Signaling Are Determined by K-Ras Activation Kinetics. Mol. Cell. Biol. 2013, 33, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 2018, 37, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, M.H.; Kim, D.W.; Lee, S.; Huang, S.; Ryu, M.J.; Kim, Y.K.; Kim, S.J.; Kim, S.J.; Hwang, J.H.; et al. Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1 pathway in papillary thyroid carcinoma. PLoS ONE 2011, 6, e16180. [Google Scholar] [CrossRef]
- Blanchard, T.G.; Lapidus, R.; Banerjee, V.; Bafford, A.C.; Czinn, S.J.; Ahmed, H.; Banerjee, A. Upregulation of RASSF1A in Colon Cancer by Suppression of Angiogenesis Signaling and Akt Activation. Cell. Physiol. Biochem. 2018, 48, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, X.; Pfeifer, G.P. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J. Biol. Chem. 2011, 286, 6253–6261. [Google Scholar] [CrossRef]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef]
- Cinar, B.; Fang, P.K.; Lutchman, M.; Di Vizio, D.; Adam, R.M.; Pavlova, N.; Rubin, M.A.; Yelick, P.C.; Freeman, M.R. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007, 26, 4523–4534. [Google Scholar] [CrossRef]
- Schmidt, M.L.; Hobbing, K.R.; Donninger, H.; Clark, G.J. RASSF1A Deficiency Enhances RAS-Driven Lung Tumorigenesis. Cancer Res. 2018, 78, 2614–2623. [Google Scholar] [CrossRef]
- Bousquet, E.; Mazieres, J.; Privat, M.; Rizzati, V.; Casanova, A.; Ledoux, A.; Mery, E.; Couderc, B.; Favre, G.; Pradines, A. Loss of RhoB expression promotes migration and invasion of human bronchial cells via activation of AKT1. Cancer Res. 2009, 69, 6092–6099. [Google Scholar] [CrossRef]
- Guo, W.; Dong, Z.; Chen, Z.; Yang, Z.; Wen, D.; Kuang, G.; Guo, Y.; Shan, B. Aberrant CpG island hypermethylation of RASSF1A in gastric cardia adenocarcinoma. Cancer Investig. 2009, 27, 459–465. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Na, A.R.; Lee, M.S.; Yoon, S.; Kim, J.S.; Yoo, Y.D. RASSF1A suppresses oncogenic H-Ras-induced c-Jun N-terminal kinase activation. Int. J. Oncol. 2006, 29, 1541–1547. [Google Scholar] [CrossRef]
- Rong, R.; Jin, W.; Zhang, J.; Sheikh, M.S.; Huang, Y. Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene 2004, 23, 8216–8230. [Google Scholar] [CrossRef]
- Song, M.S.; Chang, J.S.; Song, S.J.; Yang, T.H.; Lee, H.; Lim, D.S. The centrosomal protein RAS association domain family protein 1A (RASSF1A)-binding protein 1 regulates mitotic progression by recruiting RASSF1A to spindle poles. J. Biol. Chem. 2005, 280, 3920–3927. [Google Scholar] [CrossRef]
- Rong, R.; Jiang, L.Y.; Sheikh, M.S.; Huang, Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007, 26, 7700–7708. [Google Scholar] [CrossRef]
- Song, S.J.; Song, M.S.; Kim, S.J.; Kim, S.Y.; Kwon, S.H.; Kim, J.G.; Calvisi, D.F.; Kang, D.; Lim, D.S. Aurora A regulates prometaphase progression by inhibiting the ability of RASSF1A to suppress APC-Cdc20 activity. Cancer Res. 2009, 69, 2314–2323. [Google Scholar] [CrossRef]
- Dulgar, O.; Kutuk, T.; Eroglu, Z. Mechanisms of Resistance to BRAF-Targeted Melanoma Therapies. Am. J. Clin. Dermatol. 2021, 22, 1–10. [Google Scholar] [CrossRef]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.S. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef]
- Aylon, Y.; Yabuta, N.; Besserglick, H.; Buganim, Y.; Rotter, V.; Nojima, H.; Oren, M. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene 2009, 28, 4469–4479. [Google Scholar] [CrossRef]
- Jimenez, A.P.; Traum, A.; Boettger, T.; Hackstein, H.; Richter, A.M.; Dammann, R.H. The tumor suppressor RASSF1A induces the YAP1 target gene ANKRD1 that is epigenetically inactivated in human cancers and inhibits tumor growth. Oncotarget 2017, 8, 88437–88452. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Drablos, F.; Rye, M.B. DNA hypermethylation associated with upregulated gene expression in prostate cancer demonstrates the diversity of epigenetic regulation. BMC Med. Genom. 2020, 13, 6. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Vyas, P.; Hajduch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [PubMed]
- Howell, P.M.; Liu, Z.; Khong, H.T. Demethylating Agents in the Treatment of Cancer. Pharmaceuticals 2010, 3, 2022–2044. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Xu, Y.H.; Chen, Q.; Tang, L.; Li, A.G.; Zhang, L.F.; Zhang, C.F.; Song, J.F.; Du, Z.Z. The loss-of-function of DNA methyltransferase 1 by siRNA impairs the growth of non-small cell lung cancer with alleviated side effects via reactivation of RASSF1A and APC in vitro and vivo. Oncotarget 2017, 8, 59301–59311. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Munck, J.; Tang, J.; Taverna, P.; Wang, Y.; Miller, D.F.; Pilrose, J.; Choy, G.; Azab, M.; Pawelczak, K.S.; et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin. Cancer Res. 2014, 20, 6504–6516. [Google Scholar] [CrossRef] [PubMed]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef]
- Reu, F.J.; Bae, S.I.; Cherkassky, L.; Leaman, D.W.; Lindner, D.; Beaulieu, N.; MacLeod, A.R.; Borden, E.C. Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J. Clin. Oncol. 2006, 24, 3771–3779. [Google Scholar] [CrossRef]
- Dammann, R.H.; Richter, A.M.; Jimenez, A.P.; Woods, M.; Kuster, M.; Witharana, C. Impact of Natural Compounds on DNA Methylation Levels of the Tumor Suppressor Gene RASSF1A in Cancer. Int. J. Mol. Sci. 2017, 18, 2160. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PATHWAY | GENE | ALTERATION | IMPLICATION | Intrinsic | Acquired | Res BRAFi | Res MEKi | Ref |
|---|---|---|---|---|---|---|---|---|
| Cell Cycle | CCND1 | High copy number | Increased expression | + | + | [41] | ||
| CCND1 | Amplification | Increased expression | + | + | [42] | |||
| CDK4 | K22Q, R24C/L | Activating mutation | + | − | [42] | |||
| CDK4 | R24C/L | Activating mutation | +/− | +/− | +/− | +/− | [43] | |
| CDKN2A | Low copy number | Low expression | + | + | [41] | |||
| CDKN2A | D84N, M53T, N71fs | Inactivating mutation | + | +/− | + | [43] | ||
| CDKN2A | deletion, truncation, missense mutation | Loss of function | +/− | +/− | +/− | +/− | [44] | |
| MAPK | NRAS | Missense | Activating mutation | + | + | [45] | ||
| NRAS | Q61 | Activating mutation | + | + | [46] | |||
| NRAS | Q61K | Activating mutation | + | + | − | [47] | ||
| CRAF | Increased levels | Increased MAPK signalling | + | + | [48] | |||
| BRAFV600E | Amplification | Increased MAPK signalling | +/− | +/− | +/− | [44] | ||
| BRAFV600E | High copy number | Increased MAPK signalling | + | + | − | [49] | ||
| BRAFV600E | Amplification | Increased MAPK signalling | + | + | [45] | |||
| BRAFV600E | Splice variant (p61BRAFV600E) | Increased MAPK signalling | + | + | [50] | |||
| MAP2K1 | P124SQ/S | Activating mutation | + | + | + | [51] | ||
| MAP2K1 | P124S | Activating mutation | +/− | +/− | [44] | |||
| MAP2K1 | Q56P | Activating mutation | +/− | +/− | [44] | |||
| MAP2K1 | Q56P, E203K | Activating mutation | + | + | [46] | |||
| MAP2K1 | P124L | Activating mutation | + | + | [43] | |||
| MAP2K1 | V60E, G128V, V154I | Activating mutation | + | + | [45] | |||
| MAP2K1 | P124S/L | Activating mutation | + | + | [45] | |||
| MAP2K2 | V35M, L46F, C125S N126D | Activating mutation | + | + | [45] | |||
| MAP2K2 | W251Ter, A182V | + | +/− | + | [43] | |||
| NF1 | X2441_splice | Loss of expression | + | + | [44] | |||
| NF1 | Gln282fs, Arg440 * | Loss of function | − | [52] | ||||
| NF1 | P195S | + | +/− | [43] | ||||
| MAP3K8 (COT) | Increased levels | Increased ERK signalling | + | + | + | + | [53] | |
| RTK | EGFR | Amplification, R451C | Increased activity | + | +/− | +/− | +/− | [43] |
| EGFR | Demethylation of EGFR regulatory DNA elements | Increased PI3K/AKT signalling | + | + | [54] | |||
| IGF-1R | Increased levels | Increased PI3K/AKT signalling | + | + | − | [55] | ||
| AXL | Increased levels | + | + | + | + | [56] | ||
| KIT | Amplification, G498S | Increased activity | + | +/− | + | + | [43] | |
| PDGFRβ | Increased levels | Independent MAPK-pro-survival | + | + | [47] | |||
| SOX10 | Low levels | Increased RTK | − | + | + | [57] | ||
| T. microenvironment | HGF | Stromal secretion | Activation of MET | + | + | [58] | ||
| PI3K/AKT | PIK3CA | V344G, E545K, H1047R | Activating mutation | +/− | +/− | [44] | ||
| PIK3CA | missense | + | + | + | [45] | |||
| PTEN | mutation, deletion | Loss of function | +/− | +/− | [41] | |||
| PTEN | missense mutation, indel | Loss of function | + | + | [45] | |||
| PTEN | missense mutation, non-sense mutation, deletion, indel | Loss of function | + | + | [45] | |||
| PTEN | Loss/deletion, splice, T27C, P244fs | Loss of function | + | + | +/− | +/− | [43] | |
| PTEN | deletions, truncation, missense mutation | Loss of function | + | + | + | [44] | ||
| Small GTPases (other than RAS) | RAC | P29S | Activating mutation | + | + | [59] | ||
| RAC | P29S | Activating mutation | + | + | [60] | |||
| RAC | P29S/L | Activating mutation | + | + | + | [61] | ||
| RAC | P29S | Activating mutation | + | + | + | [43] | ||
| RAC | P29S | Activating mutation | + | [62] | ||||
| RAC | P29S | Activating mutation | + | + | [45] | |||
| Metabolic rewiring | MITF | Amplification | Increased activity | + | + | + | [45] | |
| MITF | Amplifications, G6R, R316K, S502F | Increased activity | + | +/− | +/− | − | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McKenna, S.; García-Gutiérrez, L. Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing? Int. J. Mol. Sci. 2021, 22, 5115. https://doi.org/10.3390/ijms22105115
McKenna S, García-Gutiérrez L. Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing? International Journal of Molecular Sciences. 2021; 22(10):5115. https://doi.org/10.3390/ijms22105115
Chicago/Turabian StyleMcKenna, Stephanie, and Lucía García-Gutiérrez. 2021. "Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing?" International Journal of Molecular Sciences 22, no. 10: 5115. https://doi.org/10.3390/ijms22105115
APA StyleMcKenna, S., & García-Gutiérrez, L. (2021). Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing? International Journal of Molecular Sciences, 22(10), 5115. https://doi.org/10.3390/ijms22105115