
KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism


{kind=link}
{kind=link}
Abstract
1. Introduction
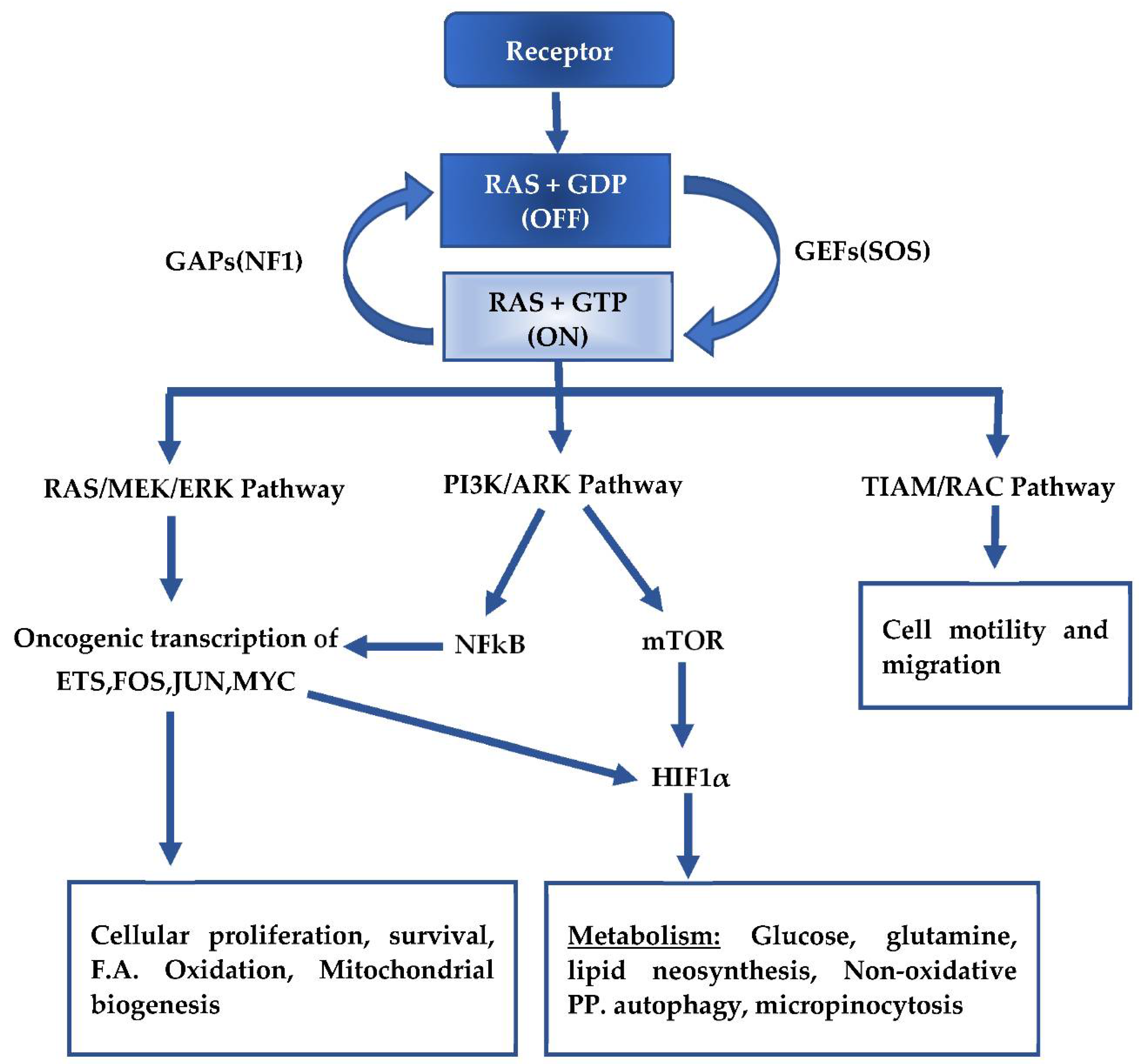
2. Mutant KRAS Signaling and Metabolism
3. Glucose Metabolism
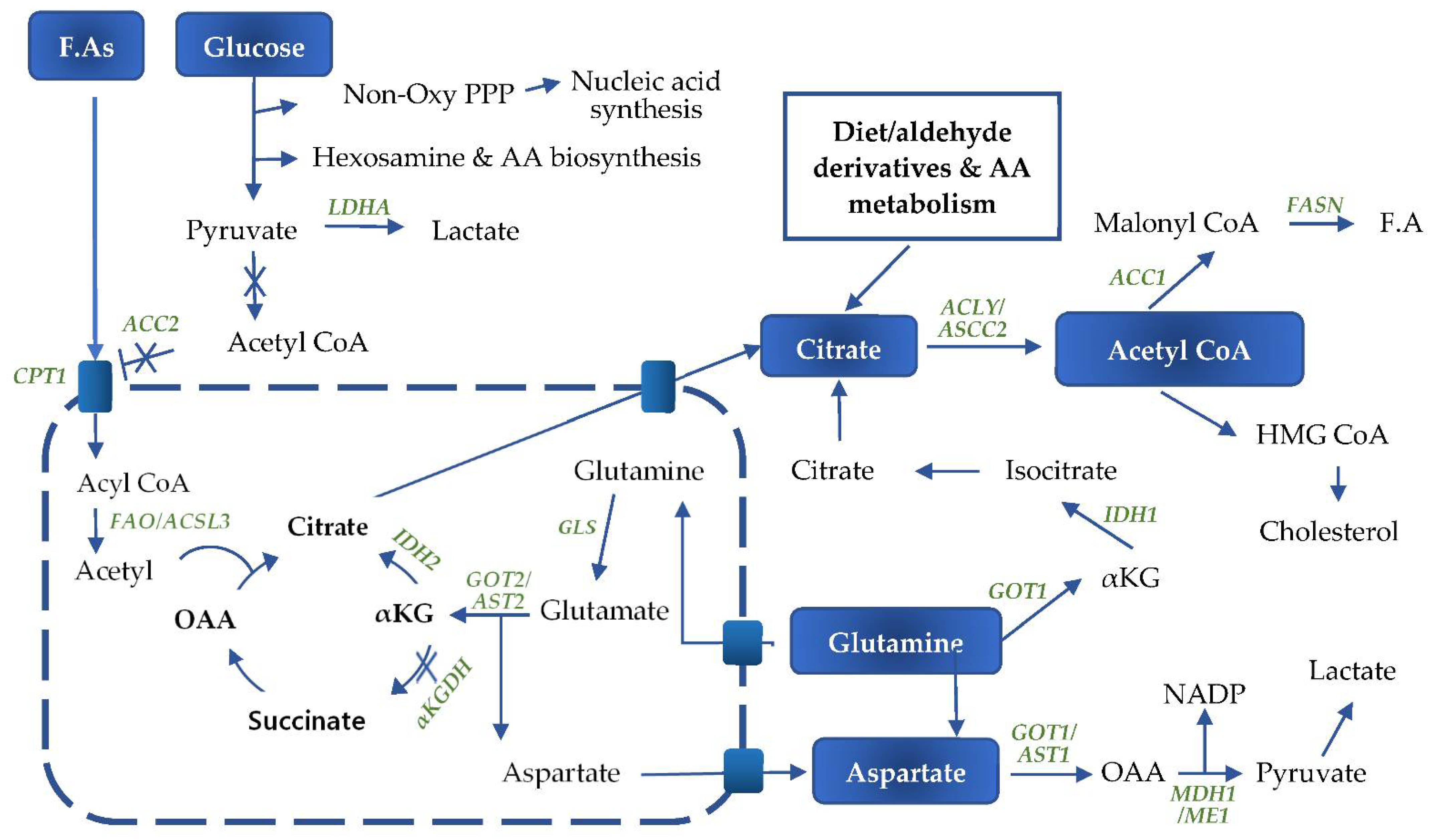
4. Glutamine Metabolism
5. Fatty Acid Metabolism
6. Fatty Acid Oxidation
7. Other Metabolic Processes
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Pancreas Fact Sheet, Globocan 2018; World Health Organization: Geneva, Switzerland, 2020; pp. 11–12. [Google Scholar]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Solomon, S.; Das, S.; Brand, R.; Whitcomb, D.C. Inherited pancreatic cancer syndromes. Cancer J. 2012, 18, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Ghiorozo, P. Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 2013, 51, 14–24. [Google Scholar]
- Lu, Q.-L.; Zhang, L.; Yee, J.K.; Go, W.V.-L.; Lee, W.-N. Metabolic consequences of LDHA inhibition by Epigallocatechin Gallate and Oxamate in MIA PaCa-2 pancreatic cancer cells. Metabolomics 2015, 11, 71–80. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.-M.; Neoptolemos, J.; Real, F.X.; van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef]
- Sapna, P.; Alexandra, E.F.; Matthias, H. Plasticity and dedifferentiation within the pancreas: Development, homeostasis, and disease. Cell Stem. Cell 2015, 16, 18–31. [Google Scholar]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P., IV; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Sharon, Y.G.F.; Gerald, C.C.; Eric, L.S.; Nomeda, G.; Gregory, D.; Denise, C.; Eliza, V.; Ronald, A.D.; Tyler, J. Context-dependent transformation of adult pancreatic cells by Oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.-M.; Yu, J.; Borgoes, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroentrology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Salvia, R.; Casciani, F.; Sereni, E.; Bassi, C. Pancreatic cancer–What’s next? Press. Med. 2019, 48, e187–e197. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Raphael, B.J. Integrated genomic characterization of pancreatic ductal adenocarcinoma the cancer genome atlas research network. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.L.; Hayward, N.K. Molecular pathways: Mitogen-activated protein kinase pathway mutations and drug resistance. Clin. Cancer Res. 2013, 19, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta Bioenerg. 2007, 1773, 1177–1195. [Google Scholar] [CrossRef]
- Ward, S.G.; Finan, P. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr. Opin. Pharm. 2003, 3, 426–434. [Google Scholar] [CrossRef]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef]
- Collins, A.; Bloomston, M. Diagnosis and management of pancreatic cancer. Minerva Gastroenterol. Dietol. 2009, 55, 445–454. [Google Scholar] [PubMed]
- Safi, F.; Roscher, R.; Bittner, R.; Schenkluhn, B.; Dopfer, H.-P.; Beger, H.G. High Sensitivity and Specificity of CA 19-9 for Pancreatic Carcinoma in Comparison to Chronic Pancreatitis. Serological and Immunohistochemical Findings. Pancreas 1987, 2, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Ii, G.V.B.; Fisher, W.E. Pancreatic cancer: Advances in treatment. World J. Gastroenterol 2014, 20, 9354–9360. [Google Scholar]
- Werner, J.; Combs, S.E.; Springfeld, C.; Hartwig, W.; Hackert, T.; Büchler, M.W. Advanced-stage pancreatic cancer: Therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef]
- Teague, A.; Lim, K.-H.; Wang-Gillam, A. Advanced pancreatic adenocarcinoma: A review of current treatment strategies and developing therapies. Ther. Adv. Med Oncol. 2015, 7, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, V.; Sperduti, I.; Milella, M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 365, 768–769. [Google Scholar]
- Gillentine, M.A.; Berry, L.N.; Goin-Kochel, R.P.; Ali, M.A.; Ge, J.; Guffey, D.; Rosenfeld, J.A.; Hanning, V.; Bader, P.; Proud, M.; et al. The cognitive and behavioral phenotypes of individuals with CHRNA7 duplications. J. Autism Dev. Disord. 2017, 47, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Boige, V.; et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized trial. J. Clin. Oncol. 2013, 31, 23–29. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. 2017, 114, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef]
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2006, 120, 1355–1363. [Google Scholar] [CrossRef]
- Duxbury, M.S.; Ito, H.; Benoit, E.; Waseem, T.; Ashley, S.W.; Whang, E.E. RNA interference demonstrates a novel role for integrin-linked kinase as a determinant of pancreatic adenocarcinoma cell gemcitabine chemoresistance. Clin. Cancer Res. 2005, 11, 3433–3438. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Büchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2017, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Kurata, N.; Ohuchida, K.; Mizumoto, K.; Mahawithitwong, P.; Sakai, H.; Onimaru, M.; Manabe, T.; Ohtsuka, T.; Tanaka, M. Predicting the chemosensitivity of pancreatic cancer cells by quantifying the expression levels of genes associated with the metabolism of gemcitabine and 5-fluorouracil. Int. J. Oncol. 2011, 39, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef] [PubMed]
- Huguet, F.; Mukherjee, S.; Javle, M. Locally advanced pancreatic cancer: The role of de fi nitive chemoradiotherapy statement of search strategies and sources of information. Clin. Oncol. 2020, 26, 560–568. [Google Scholar] [CrossRef]
- Roeder, F. Neoadjuvant radiotherapeutic strategies in pancreatic cancer. World J. Gastrointest. Oncol. 2016, 8, 186–197. [Google Scholar] [CrossRef]
- Brunner, T.B.; Scott-Brown, M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat. Oncol. 2010, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Quintiliani, M. Modification of radiation sensitivity: The oxygen effect. Int. J. Radiation Oncology Biol. Phys. 1979, 5, 1069–1076. [Google Scholar] [CrossRef]
- Matsumoto, S.; Kishimoto, S.; Saito, K.; Takakusagi, Y.; Devasahayam, N.; Hart, C.P.; Gillies, R.J.; Munasinghe, J.P.; Mitchell, J.B.; Krishna, M.C. Metabolic and Physiologic Imaging Biomarkers of the Tumor Microenvironment Predict Treatment Outcome with Radiation or a Hypoxia-Activated Prodrug in Mice. Cancer Res. 2018, 78, 3783–3792. [Google Scholar] [CrossRef]
- Mathews, L.A.; Cabarcas, S.M.; Hurt, E.M.; Zhang, X.; Jaffee, E.M.; Farrar, W.L. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas 2011, 40, 730–739. [Google Scholar] [CrossRef]
- Wang, F.; Xia, X.; Yang, C.; Shen, J.; Mai, J.; Kim, H.-C.; Kirui, D.; Kang, Y.; Fleming, J.B.; Koay, E.J.; et al. Eugene J KoaySMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin. Cancer Res. 2018, 24, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Eke, I.; Storch, K.; Haase, M.; Baretton, G.B.; Cordes, N. Caveolin-1 mediated radioresistance of 3D grown pancreatic cancer cells. Radiother. Oncol. 2009, 92, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nat. Cell Biol. 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2015, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef]
- Deley, D.; Zambirinis, C.P.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, V.R.K.; et al. γδ T Cells Support Pancreatic Oncogenesis by Restraining αβ T Cell Activation. Cell 2016, 166, 1485–1499. [Google Scholar] [CrossRef]
- Hu, H.; Hang, J.-J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.-W. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumor Biol. 2016, 37, 8657–8664. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III Study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest oncology group–directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Jung, K.; Baek, D.-S.; Hong, S.-S. Co-targeting of EGF receptor and neuropilin-1 overcomes cetuximab resistance in pancreatic ductal adenocarcinoma with integrin β1-driven Src-Akt bypass signaling. Oncogene 2016, 36, 2543–2552. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase iii trial of the national cancer institute of canada clinical trials group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Ormanns, S.; Haas, M.; Modest, D.P.; Kirchner, T.; et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J. Gastroenterol. 2013, 48, 544–548. [Google Scholar] [CrossRef]
- Conradt, L.; Godl, K.; Schaab, C.; Tebbe, A.; Eser, S.; Diersch, S.; Michalski, C.W.; Kleeff, J.; Schnieke, A.; Schmid, R.M.; et al. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia 2011, 13, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.B.; Shen, G.-L.; Holloway, S.E.; Davis, M.; Brekken, R.A. Molecular consequences of silencing mutant k-ras in pancreatic cancer cells: Justification for k-ras–directed therapy. Mol. Cancer Res. 2005, 3, 413–423. [Google Scholar] [CrossRef]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for cancer treatment: The search continues. Futur. Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.S.-H.K.; Kim, K.-W. Hypoxia-inducible-factor-hif--its-p. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kaelin, W.G. The von Hippel-Lindau tumor suppressor gene. Exp. Cell Res. 2001, 264, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem 2005, 74, 115–128. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Briggs, J.; Deng, J.; Ma, J.; Lee, H.; Kortylewski, M.; Kujawski, M.; Kay, H.; Cress, W.D.; Jove, R.; et al. Signal Transducer and Activator of Transcription 3 is required for hypoxia-inducible factor-1α RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res. 2008, 6, 1099–1105. [Google Scholar] [CrossRef]
- Laughner, E.; Taghavi, P.; Chiles, K.; Patrick, C.; Semenza, G.L.; Mahon, P.C. HER2 (neu) signaling increases the rate of synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Am. Soc. Microbiol. 2001, 21, 3995–4004. [Google Scholar]
- Brix, B.; Mesters, J.R.; Pellerin, L.; Jöhren, O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1α-mediated target gene activation. J. Neurosci. 2012, 32, 9729–9735. [Google Scholar] [CrossRef]
- Brahimi-Horn, C.; Pouysségur, J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bull Cancer 2006, 93, E73–E80. [Google Scholar]
- Moench, R.; Grimmig, T.; Kannen, V.; Tripathi, S.; Faber, M.; Moll, E.-M.; Chandraker, A.; Lissner, R.; Germer, C.-T.; Waaga-Gasser, A.M.; et al. Exclusive inhibition of PI3K/Akt/mTOR signaling is not sufficient to prevent PDGF-mediated effects on glycolysis and proliferation in colorectal cancer. Oncotarget 2016, 7, 68749–68767. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Costas, A.; Lyssiotis, C.A.; Hua, S.; Chu, G.C. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Moon, J.-S.; Jin, W.-J.; Kwak, J.-H.; Kim, H.-J.; Yun, M.-J.; Kim, J.-W.; Park, S.W.; Kim, K.-S. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem. J. 2010, 433, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Gohar, A.; Zarei, M.; Brody, J.R.; Winter, J.M. Metabolic Dependencies in Pancreatic Cancer. Front. Oncol. 2018, 8, 617. [Google Scholar] [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.C.; Trenary, I.A.; Young, J.D.; Li, M.; Lieber, D.C. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.-A.; Initiative, A.P.C.G.; van Dam, E.; Cowley, M.; Han, T.-L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 1–15. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2008, 458, 762–765. [Google Scholar] [CrossRef]
- Bott, A.J.; Maimouni, S.; Zong, W.-X. The pleiotropic effects of glutamine metabolism in cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nat. Cell Biol. 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 2019, 29, 1287–1298.e6. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, B.J.A.Z.E.S.C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Perera, R.M.; Bardeesy, N. Pancreatic cancer metabolism: Breaking it down to build it back up. Cancer Discov. 2015, 5, 1247–1261. [Google Scholar] [CrossRef]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2015, 3, e1033586. [Google Scholar] [CrossRef]
- He, Y.; Hakvoort, T.B.M.; Köhler, S.E.; Vermeulen, J.L.M.; de Waart, D.R.; de Theije, C.; Have, G.A.M.T.; van Eijk, H.M.H.; Kunne, C.; Labruyere, W.T.; et al. Glutamine Synthetase in Muscle Is Required for Glutamine Production during Fasting and Extrahepatic Ammonia Detoxification. J. Biol. Chem. 2010, 285, 9516–9524. [Google Scholar] [CrossRef]
- Souba, W.W.; Herskowitz, K.D.; Plumley, D.A. Lung glutamine metabolism. JPEN J. Parenter. Enteral. Nutr. 1990, 14, 68S–70S. [Google Scholar] [CrossRef]
- Moreadiths, R.W.; Lehningert, A.L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 1984, 259, 6215–6221. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis that Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Zhou, W.; Wang, J.; Huang, X.; Zuo, Y.; Wang, T.-S.; Gao, X.; Xu, Y.-Y.; Zou, S.-W.; Liu, Y.-B.; et al. Arginine Methylation of MDH1 by CARM1 Inhibits Glutamine Metabolism and Suppresses Pancreatic Cancer. Mol. Cell 2016, 64, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nat. Cell Biol. 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Son, J.; Cantley, L.C.; Kimmelman, A.C. Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle 2013, 12, 1987–1988. [Google Scholar] [CrossRef]
- Duan, G.; Shi, M.; Xie, L.; Xu, M.; Wang, Y.; Yan, H.; Zhuge, Y.; Zou, X. Increased Glutamine Consumption in Cisplatin-Resistant Cells Has a Negative Impact on Cell Growth. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Serpa, J. Metabolic cooperation between cancer and non-cancerous stromal cells is pivotal in cancer progression. Tumor Biol. 2018, 40, 1–15. [Google Scholar] [CrossRef]
- Izuishi, K.; Kato, K.; Ogura, T.; Kinoshita, T.; Esumi, H. Remarkable tolerance of tumor cells to nutrient deprivation: Possible new biochemical target for cancer therapy. Cancer Res. 2000, 60, 6201–6207. [Google Scholar]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; Broek, N.J.F.V.D.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nat. Cell Biol. 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Nishi, K.; Suzuki, M.; Yamamoto, N.; Matsumoto, A.; Iwase, Y.; Yamasaki, K.; Otagiri, M.; Yumita, N. Glutamine deprivation enhances Acetyl-CoA carboxylase inhibitor-induced death of human pancreatic cancer cells. Anticancer Res. 2018, 38, 6683–6689. [Google Scholar] [CrossRef] [PubMed]
- Margittai, É.; Bánhegyi, G. Isocitrate dehydrogenase: A NADPH-generating enzyme in the lumen of the endoplasmic reticulum. Arch. Biochem. Biophys. 2008, 471, 184–190. [Google Scholar] [CrossRef]
- Brody, J.R.; Yabar, C.S.; Zarei, M.; Bender, J.; Matrisian, L.M.; Rahib, L.; Heartwell, C.; Mason, K.; Yeo, C.J.; Peiper, S.C.; et al. Identification of a novel metabolic-related mutation (IDH1) in metastatic pancreatic cancer. Cancer Biol. Ther. 2018, 19, 249–253. [Google Scholar] [CrossRef]
- Zhang, F.; Du, G. Dysregulated lipid metabolism in cancer. World J. Biol. Chem. 2012, 3, 167–174. [Google Scholar] [CrossRef]
- Clerc, P.; Bensaadi, N.; Pradel, P.; Estival, A.; Clemente, F.; Vaysse, N. Lipid-dependent proliferation of pancreatic cancer cell lines. Cancer Res. 1991, 51, 3633–3638. [Google Scholar] [PubMed]
- Yu, M.; Liu, H.; Duan, Y.; Zhang, D.; Li, S.; Wang, F. Four types of fatty acids exert differential impact on pancreatic cancer growth. Cancer Lett. 2015, 360, 187–194. [Google Scholar] [CrossRef]
- Singh, A.; Ruiz, C.; Bhalla, K.; Haley, J.A.; Li, Q.K.; Acquaah-Mensah, G.; Montal, E.; Sudini, K.R.; Skoulidis, F.; Wistuba, I.I.; et al. De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. FASEB J. 2018, 32, 7018–7027. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Hong, S.-M.; Nyhan, S.; Canto, M.; Fedarko, N.; Klein, A.; Griffith, M.; Omura, N.; Medghalchi, S.; Kuhajda, F.; et al. Serum Fatty Acid Synthase as a Marker of Pancreatic Neoplasia. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2380–2385. [Google Scholar] [CrossRef]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, S138–S143. [Google Scholar] [CrossRef]
- Baquet, A.; Gaussin, V.; Bollen, M.; Stalmans, W.; Hue, L. Mechanism of activation of liver acetyl-CoA carboxylase by cell swelling. Eur. J. Biochem. 1993, 217, 1083–1089. [Google Scholar] [CrossRef]
- Tadros, S.B.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Lyudmyla, B.; Lazenby, A.J.; Berim, L.; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, W.; Luo, M.; Zhou, Y.; Chang, G.; Ren, W.; Wu, K.; Guilin, C.; Shen, J.; Zhao, X.; et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumor Biol 2015, 36, 4133–4141. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Simpson, P.J.; McFadden, J.M.; Townsend, C.A.; Medghalchi, S.M.; Vadlamudi, A.; Pinn, M.L.; Ronnett, G.V.; Kuhajda, F.P. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 2003, 63, 7330–7337. [Google Scholar]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-a is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar] [PubMed]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty acid oxidation mediated by Acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Duan, J.-J.; Cai, J.; Guo, Y.-F.; Bian, X.-W.; Yu, S.-C. ALDH1A3, a metabolic target for cancer diagnosis and therapy. Int. J. Cancer 2016, 139, 965–975. [Google Scholar] [CrossRef]
- Jia, J.; Parikh, H.; Xiao, W.; Hoskins, J.W.; Pflicke, H.; Liu, X.; Collins, I.; Zhou, W.; Wang, Z.; Powell, J.; et al. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med. Genom. 2013, 6, 33. [Google Scholar] [CrossRef]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 1977, 60, 265–270. [Google Scholar] [CrossRef]
- Brownsey, R.W.; Denton, R.M. 5 Acetyl-Coenzyme A Carboxylase. Enzymes 1987, 18, 123–146. [Google Scholar]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. c-Myc programs fatty acid metabolism and dictates acetyl-coa abundance and fate. J. Bio. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2012, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, N.; van Houten, B.; Vockley, J.; et al. Mitochondrial Structure, Function and Dynamics Are Temporally Controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Wang, X. RAS: Striking at the core of the oncogenic circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Shobe, J.L.; Zhou, M.; Huang, S.; Shuman, T.; Cai, D.J.; Golshani, P.; Kamata, M.; Silva, A.J. CREB regulates memory allocation in the insular cortex Yoshitake. Curr. Biol. 2013, 498, 104–108. [Google Scholar]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muyinda, I.J.; Park, J.-G.; Jang, E.-J.; Yoo, B.-C. KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism. Int. J. Mol. Sci. 2021, 22, 5070. https://doi.org/10.3390/ijms22105070
Muyinda IJ, Park J-G, Jang E-J, Yoo B-C. KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism. International Journal of Molecular Sciences. 2021; 22(10):5070. https://doi.org/10.3390/ijms22105070
Chicago/Turabian StyleMuyinda, Isaac James, Jae-Gwang Park, Eun-Jung Jang, and Byong-Chul Yoo. 2021. "KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism" International Journal of Molecular Sciences 22, no. 10: 5070. https://doi.org/10.3390/ijms22105070
APA StyleMuyinda, I. J., Park, J.-G., Jang, E.-J., & Yoo, B.-C. (2021). KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism. International Journal of Molecular Sciences, 22(10), 5070. https://doi.org/10.3390/ijms22105070