
MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
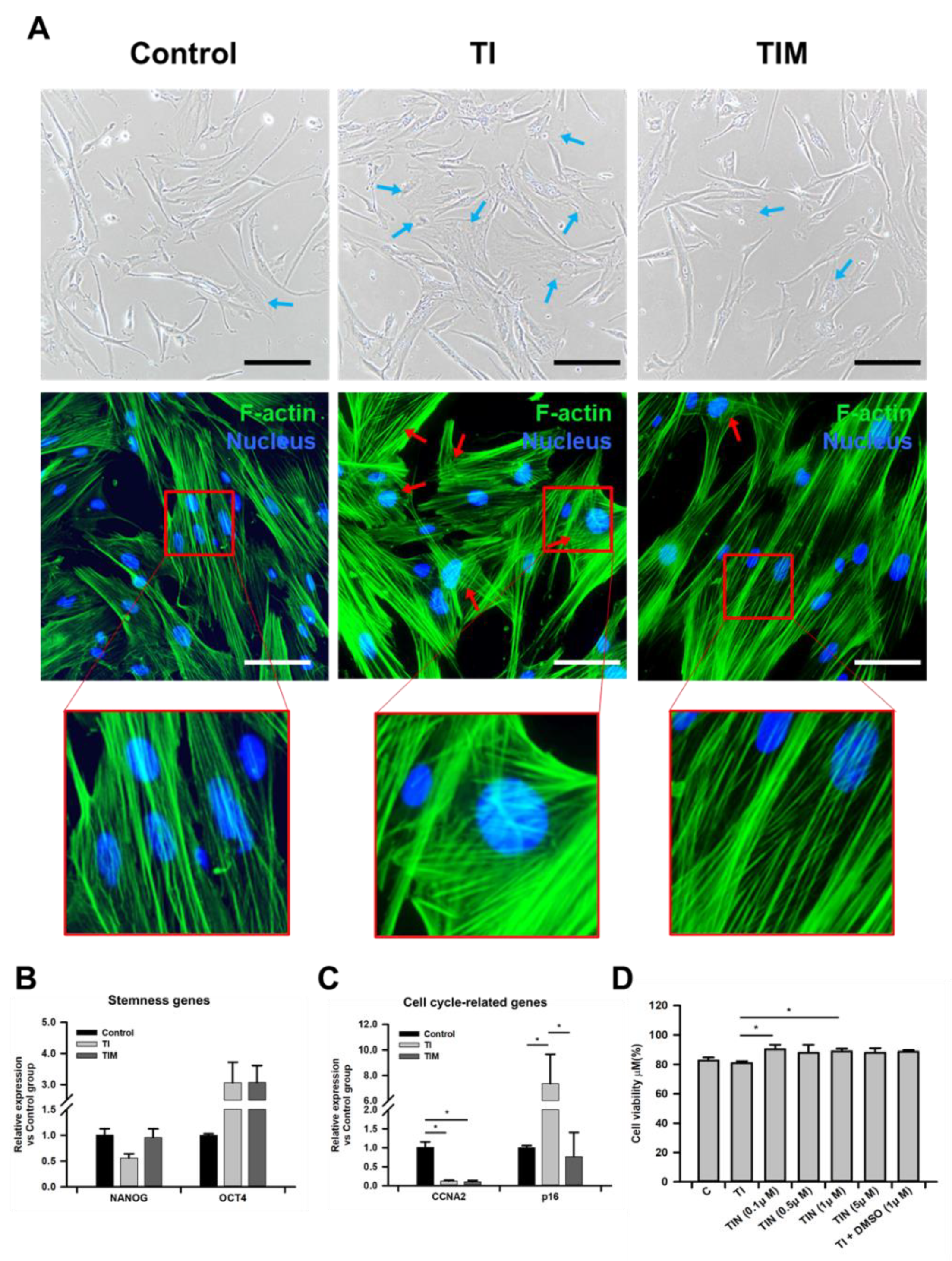
2.1. MIT-001 Ameliorates the Morphology of TNF-α/IFN-γ-Exposed hPD-MSCs
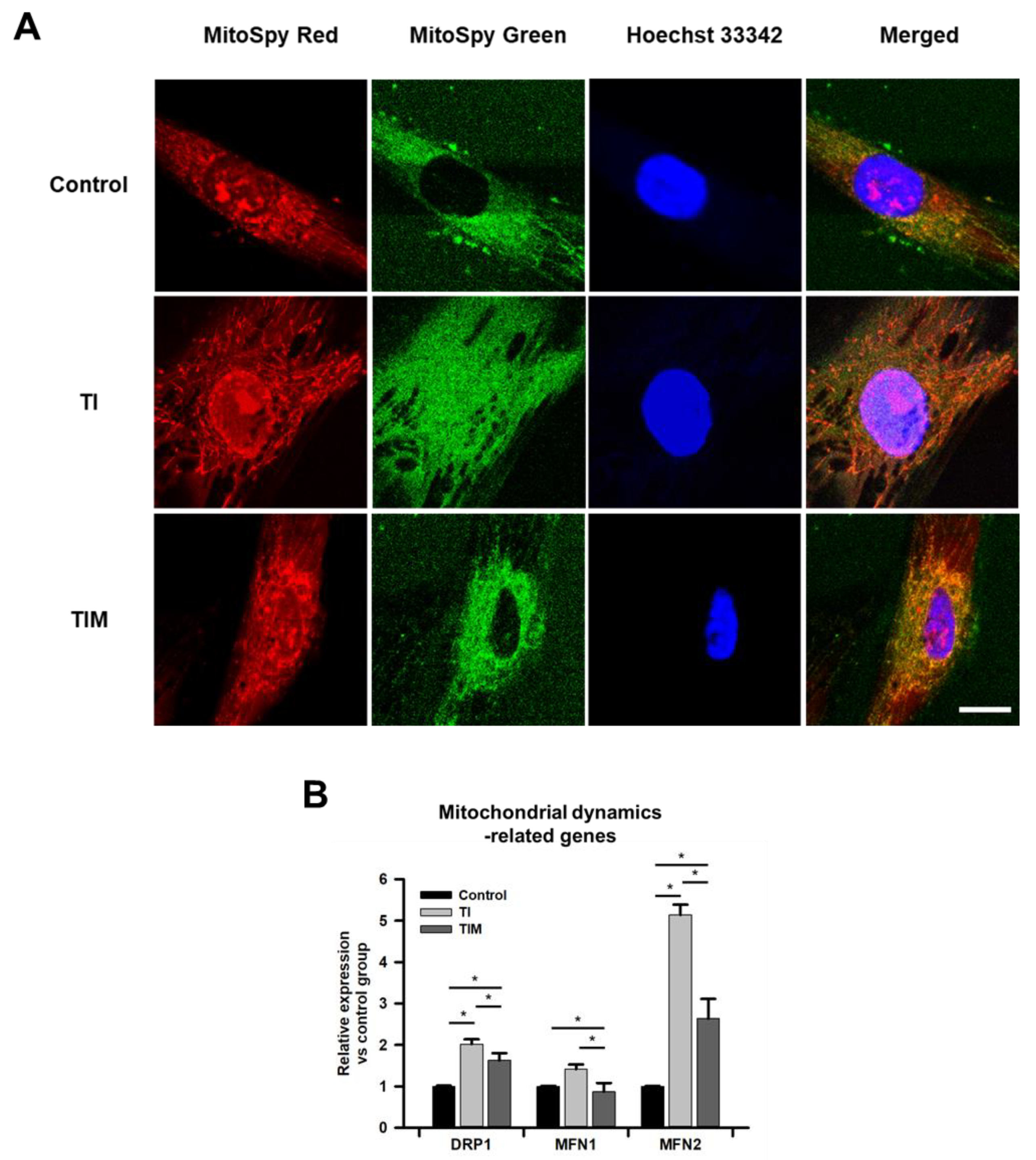
2.2. MIT-001 Remedies the Balance of the Mitochondrial Distribution and Dynamics of TNF-α/IFN-γ-Exposed hPD-MSCs
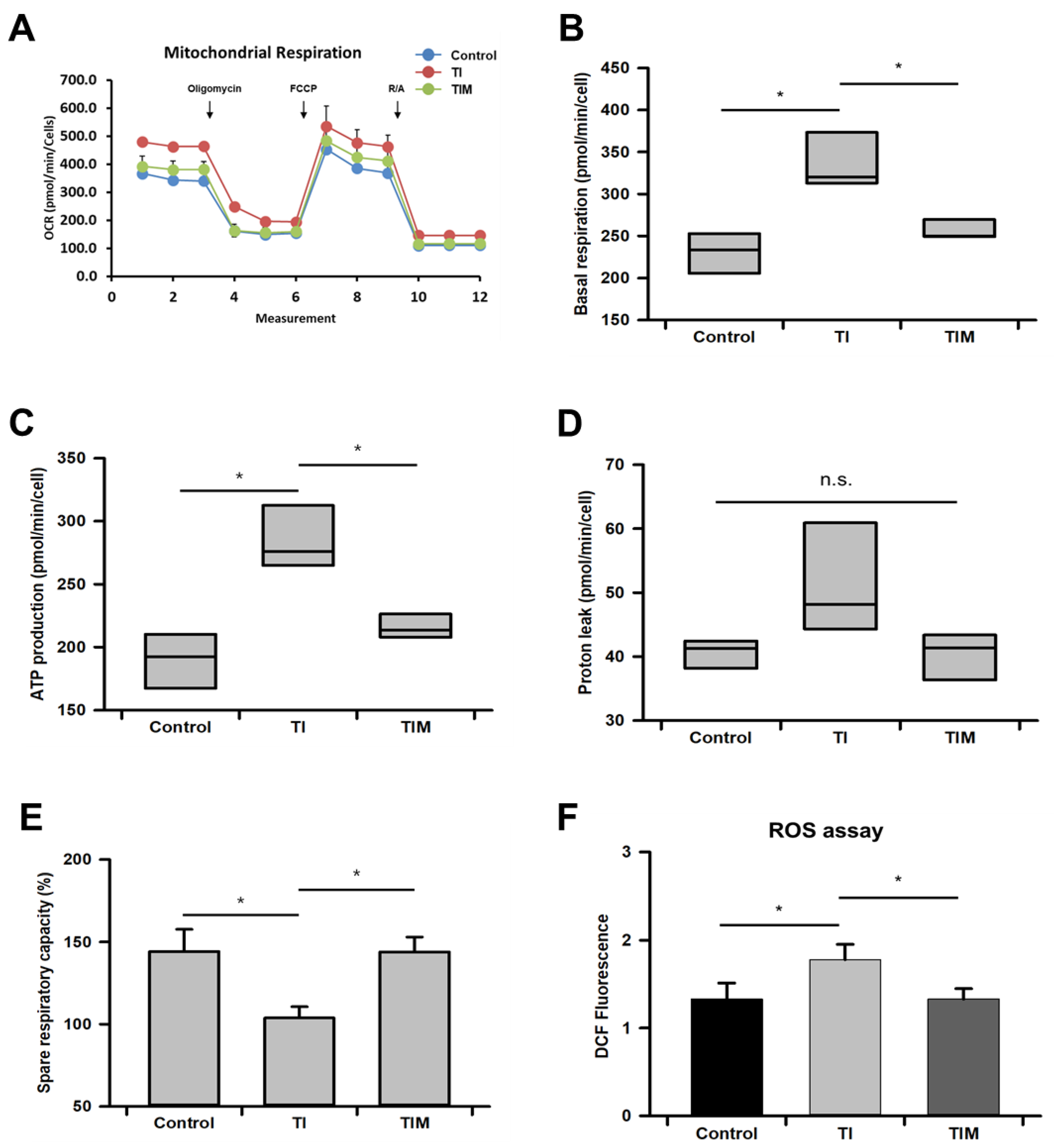
2.3. MIT-001 Reduced Mitochondrial Oxidative Stress
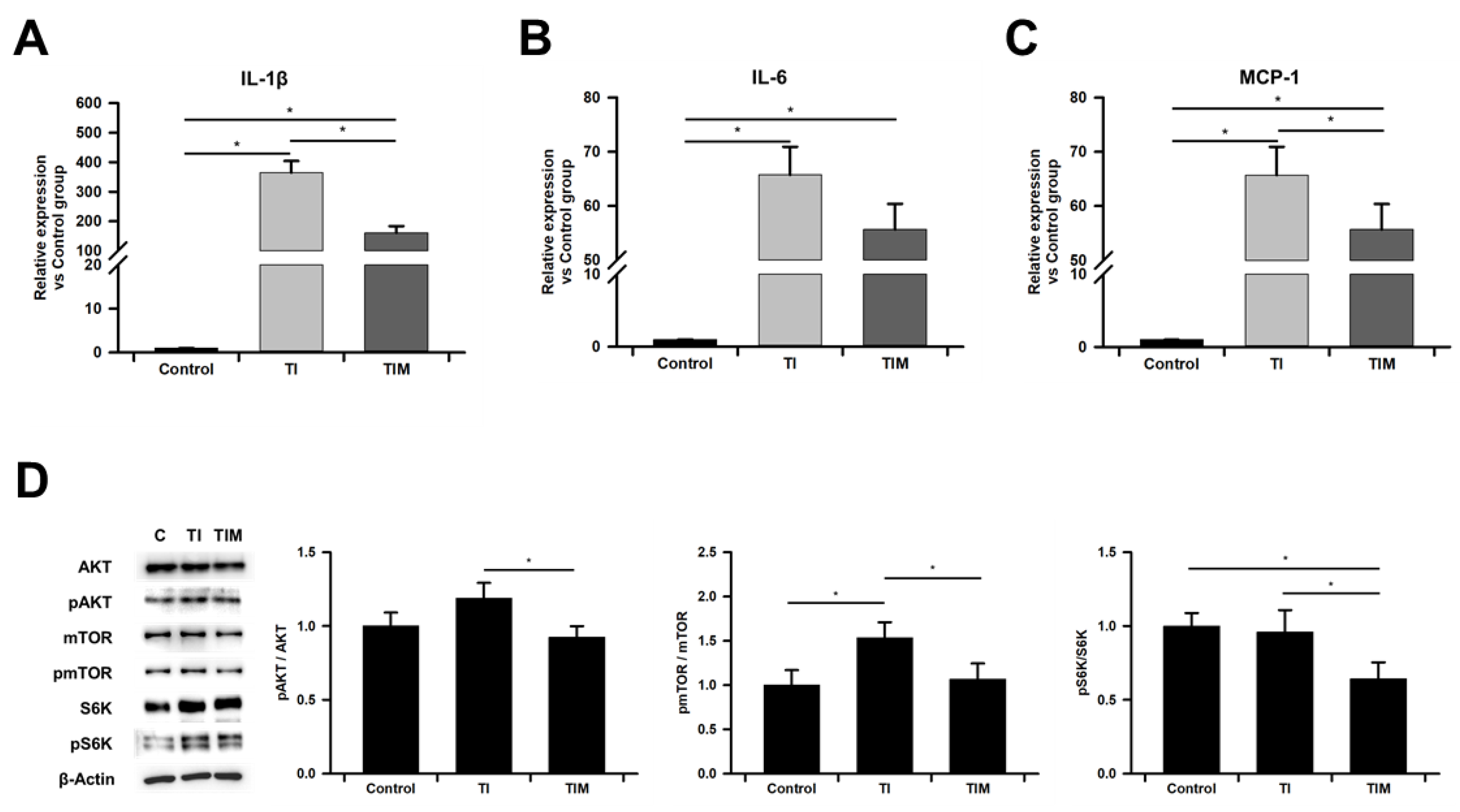
2.4. MIT-001 Ameliorated Inflammatory Phenotype of TNF-α/IFN-γ-Exposed hPD-MSCs
3. Discussion
4. Materials and Methods
4.1. Cell Culture and MIT-001 Treatment
4.2. Fluorescence Imaging of Phalloidin-Stained Cells
4.3. Reverse Transcription and Real-Time qPCR
4.4. Western Blotting
4.5. Oxygen Consumption Rate (OCR) Measurements of Cells
4.6. Mitochondrial Function Analysis of Cells
4.7. Reactive Oxygen Species (ROS) Assay
4.8. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenes-cence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Huang, J.; Zhang, H.; Wang, Y.; Matesic, L.E.; Takahata, M.; Awad, H.; Chen, D.; Xing, L. Tumor necrosis factor inhibits mesenchymal stem cell differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1. Stem Cells 2011, 29, 1601–1610. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Liu, Y.; Akiyama, K.; Chen, C.; Qu, C.; Jin, Y.; Shi, S. IFN-γ and TNF-α Synergistically Induce Mesenchymal Stem Cell Impairment and Tumorigenesis Via NFκB Signaling. Stem Cells 2013, 31, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated im-munosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Saldana, L.; Bensiamar, F.; Valles, G.; Mancebo, F.J.; Garcia-Rey, E.; Vilaboa, N. Immunoregulatory potential of mesen-chymal stem cells following activation by macrophage-derived soluble factors. Stem Cell Res. Ther. 2019, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Sakata, H.; Narasimhan, P.; Niizuma, K.; Maier, C.M.; Wakai, T.; Chan, P.H. Interleukin 6-preconditioned neural stem cells reduce ischaemic injury in stroke mice. Brain 2012, 135, 3298–3310. [Google Scholar] [CrossRef]
- Fatt, M.; Hsu, K.; He, L.; Wondisford, F.; Miller, F.D.; Kaplan, D.R.; Wang, J. Metformin Acts on Two Different Molecu-lar Pathways to Enhance Adult Neural Precursor Proliferation/Self-Renewal and Differentiation. Stem Cell Rep. 2015, 5, 988–995. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Au-tophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Khorraminejad-Shirazi, M.; Farahmandnia, M.; Kardeh, B.; Estedlal, A.; Kardeh, S.; Monabati, A. Aging and stem cell therapy: AMPK as an applicable pharmacological target for rejuvenation of aged stem cells and achieving higher effica-cy in stem cell therapy. Hematol. Oncol. Stem Cell Ther. 2018, 11, 189–194. [Google Scholar] [CrossRef]
- Das, R.; Jahr, H.; van Osch, G.J.; Farrell, E. The Role of Hypoxia in Bone Marrow–Derived Mesenchymal Stem Cells: Con-siderations for Regenerative Medicine Approaches. Tissue Eng. Part B Rev. 2010, 16, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, T. Metabolic regulation of mesenchymal stem cell in expansion and therapeutic application. Biotechnol. Prog. 2014, 31, 468–481. [Google Scholar] [CrossRef]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef]
- Gu, Y.; Li, T.; Ding, Y.; Sun, L.; Tu, T.; Zhu, W.; Hu, J.; Sun, X. Changes in mesenchymal stem cells following long-term culture in vitro. Mol. Med. Rep. 2016, 13, 5207–5215. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, Z.; Huang, S.; Wu, Y.; Liu, L.; Lin, L.; Wang, D.; He, Q.; Ruan, D. Age-Related Changes in Nucleus Pul-posus Mesenchymal Stem Cells: An In Vitro Study in Rats. Stem Cells Int. 2017, 2017, 6761572. [Google Scholar] [CrossRef]
- Romieu-Mourez, R.; François, M.; Boivin, M.-N.; Bouchentouf, M.; Spaner, D.E.; Galipeau, J. Cytokine Modulation of TLR Expression and Activation in Mesenchymal Stromal Cells Leads to a Proinflammatory Phenotype. J. Immunol. 2009, 182, 7963–7973. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Yang, R.; Chen, W.; Wang, S.; Shi, S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Zhao, Y.; Liu, H.; Chen, J.; Ren, J.; Jin, J.; Bedognetti, D.; Liu, S.; Wang, E.; Marincola, F.; et al. Interfer-on-gamma and Tumor Necrosis Factor-alpha Polarize Bone Marrow Stromal Cells Uniformly to a Th1 Phenotype. Sci. Rep. 2016, 6, 26345. [Google Scholar] [CrossRef]
- Iguchi, M.; Hiroi, M.; Kanegae, H.; Ohmori, Y. Costimulation of Murine Osteoblasts with Interferon-gamma and Tumor Necrosis Factor-alpha Induces Apoptosis through Downregulation of Bcl-2 and Release of Cytochrome c from Mito-chondria. Mediat. Inflamm. 2018, 2018, 3979606. [Google Scholar] [CrossRef] [PubMed]
- Yourek, G.; Hussain, M.A.; Mao, J.J. Cytoskeletal Changes of Mesenchymal Stem Cells during Differentiation. ASAIO J. 2007, 53, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Saidova, A.A.; Vorobjev, I.A. Lineage Commitment, Signaling Pathways, and the Cytoskeleton Systems in Mesenchymal Stem Cells. Tissue Eng. Part B: Rev. 2020, 26, 13–25. [Google Scholar] [CrossRef]
- Rodríguez, J.P.; González, M.; Ríos, S.; Cambiazo, V. Cytoskeletal organization of human mesenchymal stem cells (MSC) changes during their osteogenic differentiation. J. Cell. Biochem. 2004, 93, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.-Y.; Shin, B.-H.; Lee, M.; Lee, S.; Heo, C.Y. NecroX-5 ameliorates inflammation by skewing macrophages to the M2 phenotype. Int. Immunopharmacol. 2019, 66, 139–145. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, K.A.; Lee, M.K.; Kim, S.H.; Lee, I.K.; Kim, S.Y. A novel small molecule, NecroX-7, inhibits osteoclast differentiation by suppressing NF-kappaB activity and c-Fos expression. Life Sci. 2012, 91, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Koo, S.Y.; Ahn, B.-H.; Park, O.; Park, D.H.; Seo, D.O.; Won, J.H.; Yim, H.J.; Kwak, H.-S.; Park, H.S.; et al. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO− scavenger. Arch. Pharmacal Res. 2010, 33, 1813–1823. [Google Scholar] [CrossRef]
- Im, K.I.; Kim, N.; Lim, J.Y.; Nam, Y.S.; Lee, E.S.; Kim, E.J.; Kim, H.J.; Kim, S.H.; Cho, S.G. The Free Radical Scavenger NecroX-7 Attenuates Acute Graft-versus-Host Disease via Reciprocal Regulation of Th1/Regulatory T Cells and Inhibi-tion of HMGB1 Release. J. Immunol. 2015, 194, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.K.; Ogando, C.R.; Wang See, C.; Chang, T.Y.; Barabino, G.A. Changes in phenotype and differentiation poten-tial of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef]
- Lin, Y.; Choksi, S.; Shen, H.-M.; Yang, Q.-F.; Hur, G.M.; Kim, Y.S.; Tran, J.H.; Nedospasov, S.A.; Liu, Z.-G. Tumor Necrosis Factor-induced Nonapoptotic Cell Death Requires Receptor-interacting Protein-mediated Cellular Reactive Oxygen Species Accumulation. J. Biol. Chem. 2004, 279, 10822–10828. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.Y.; Kim, S.-Y.; Jung, K.; Cho, M.-L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nat. Cell Biol. 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and phar-macological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Borzì, R.M. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules 2020, 10, 340. [Google Scholar] [CrossRef]
- Lloyd, A.C. The Regulation of Cell Size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Giannoudis, P.V.; Burska, A.N.; Ponchel, F.; Jones, E.A. Age-related Changes in Bone Mar-row Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development. Cell Transplant. 2017, 26, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 9757201. [Google Scholar] [CrossRef] [PubMed]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.S.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef]
- Yan, W.; Diao, S.; Fan, Z. The role and mechanism of mitochondrial functions and energy metabolism in the function regulation of the mesenchymal stem cells. Stem Cell Res. Ther. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Stab, B.R., 2nd; Martinez, L.; Grismaldo, A.; Lerma, A.; Gutierrez, M.L.; Barrera, L.A.; Sutachan, J.J.; Albarracin, S.L. Mitochondrial Functional Changes Characterization in Young and Senescent Human Adipose Derived MSCs. Front Aging Neurosci. 2016, 8, 299. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.P.; Sridharan, G.V.; Hayden, R.S.; Kaplan, D.L.; Lee, K.; Georgakoudi, I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 2013, 3, 3432. [Google Scholar] [CrossRef] [PubMed]
- Anesti, V.; Scorrano, L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta. BBA Bioenerg. 2006, 1757, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L. Mitochondrial-cytoskeletal interactions: Dynamic associations that facilitate network function and remodeling. Curr. Opin. Physiol. 2018, 3, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mi-tochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef]
- Mancini, O.K.; Lora, M.; Cuillerier, A.; Shum-Tim, M.; Hamdy, R.C.; Burelle, Y.; Servant, M.J.; Stochaj, U.; Colmegna, I. Mitochondrial Oxidative Stress Reduces the Immunopotency of Mesenchymal Stromal Cells in Adults With Coronary Artery Disease. Circ. Res. 2018, 122, 255–266. [Google Scholar] [CrossRef]
- Ye, G.; Xie, Z.; Zeng, H.; Wang, P.; Li, J.; Zheng, G.; Wang, S.; Cao, Q.; Li, M.; Liu, W.; et al. Oxidative stress-mediated mitochondrial dysfunction facilitates mesenchymal stem cell senescence in ankylos-ing spondylitis. Cell Death Dis. 2020, 11, 775. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Takami, T.; Okada, S.; Hara, K.; Matsumoto, T.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Analysis of Metabolomic Changes in Mesenchymal Stem Cells on Treatment with Desferrioxamine as a Hypoxia Mimetic Compared with Hypoxic Conditions. Stem Cells 2018, 36, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, B.; Farzadi, S.; Ghuman, M.; Hughes, F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesen-chymal stem cells. Stem Cells 2014, 32, 2256–2266. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, G.; Wu, L.; Tang, Y.; Guo, C. Inhibition of ER stress-activated JNK pathway attenuates TNF-α-induced inflammatory response in bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2021, 541, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.D.; Kim, Y.J.; Cho, M.J.; Seok, J.; Kim, G.J.; Lee, C.-H.; Ko, J.J.; Kim, Y.S.; Lee, J.H. The mitochondrial-derived peptide MOTS-c promotes homeostasis in aged human placenta-derived mesenchymal stem cells in vitro. Mitochondrion 2021, 58, 135–146. [Google Scholar] [CrossRef]
- Park, J.-H.; Kim, H.K.; Jung, H.; Kim, K.H.; Kang, M.S.; Hong, J.H.; Yu, B.C.; Park, S.; Seo, S.-K.; Choi, I.W.; et al. NecroX-5 prevents breast cancer metastasis by AKT inhibition via reducing intracellular calcium levels. Int. J. Oncol. 2016, 50, 185–192. [Google Scholar] [CrossRef] [PubMed][Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.D.; Kim, Y.J.; Cho, M.J.; Kim, G.J.; Kim, S.H.; Kim, M.J.; Ko, J.J.; Lee, J.H. MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization. Int. J. Mol. Sci. 2021, 22, 5062. https://doi.org/10.3390/ijms22105062
Yu WD, Kim YJ, Cho MJ, Kim GJ, Kim SH, Kim MJ, Ko JJ, Lee JH. MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization. International Journal of Molecular Sciences. 2021; 22(10):5062. https://doi.org/10.3390/ijms22105062
Chicago/Turabian StyleYu, Won Dong, Yu Jin Kim, Min Jeong Cho, Gi Jin Kim, Soon Ha Kim, Myung Joo Kim, Jung Jae Ko, and Jae Ho Lee. 2021. "MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization" International Journal of Molecular Sciences 22, no. 10: 5062. https://doi.org/10.3390/ijms22105062
APA StyleYu, W. D., Kim, Y. J., Cho, M. J., Kim, G. J., Kim, S. H., Kim, M. J., Ko, J. J., & Lee, J. H. (2021). MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization. International Journal of Molecular Sciences, 22(10), 5062. https://doi.org/10.3390/ijms22105062