Ordinary Gasoline Emissions Induce a Toxic Response in Bronchial Cells Grown at Air-Liquid Interface

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
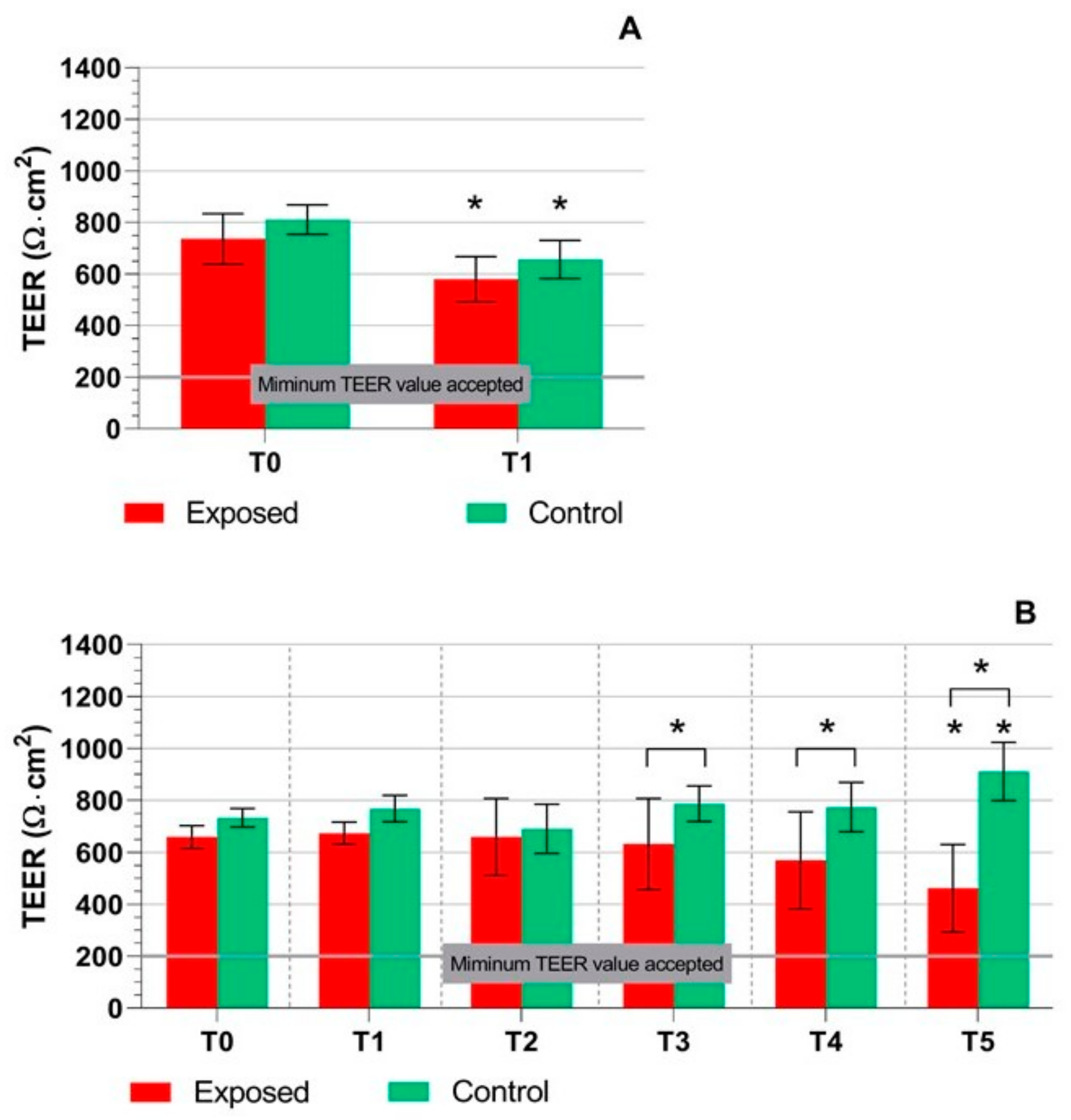
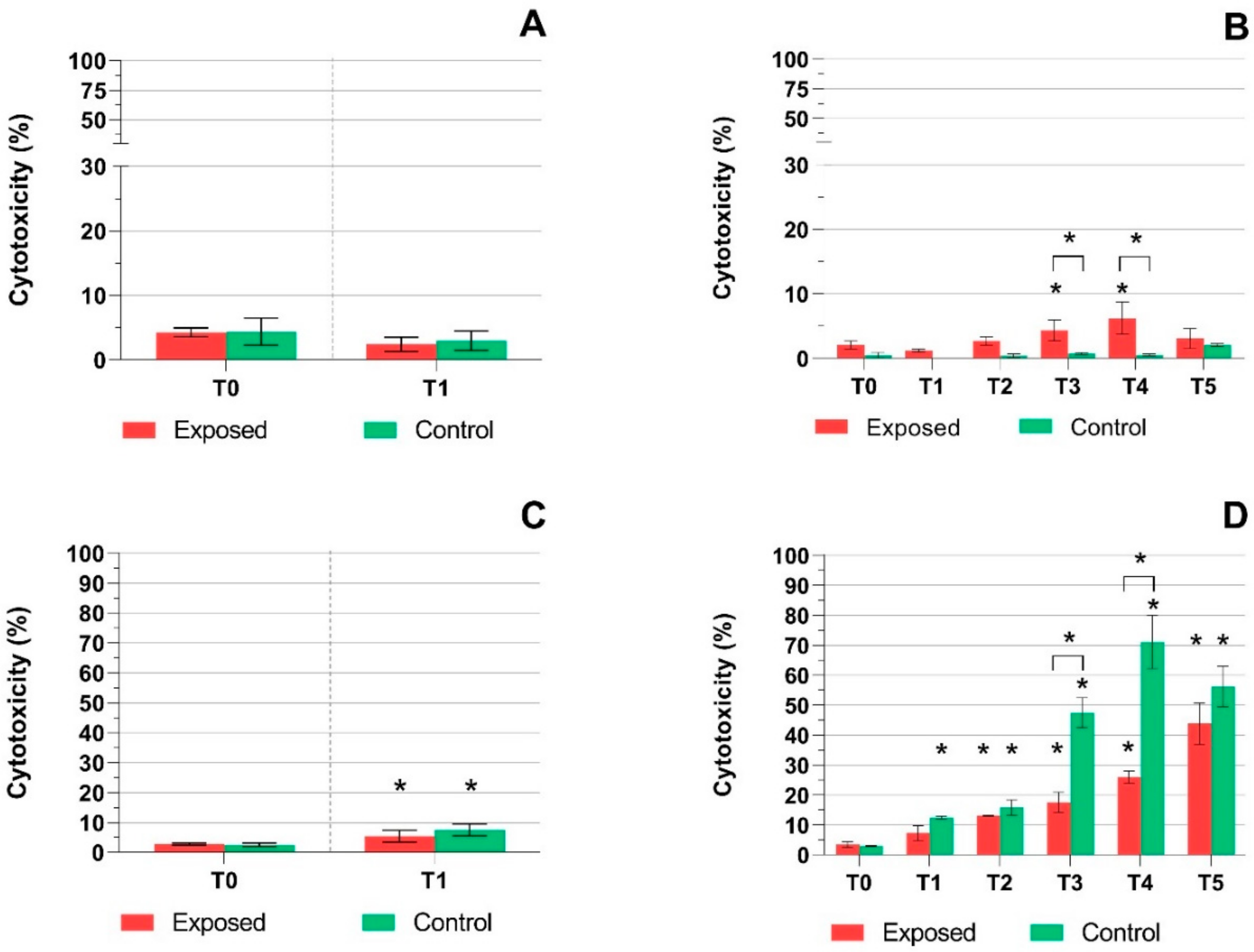
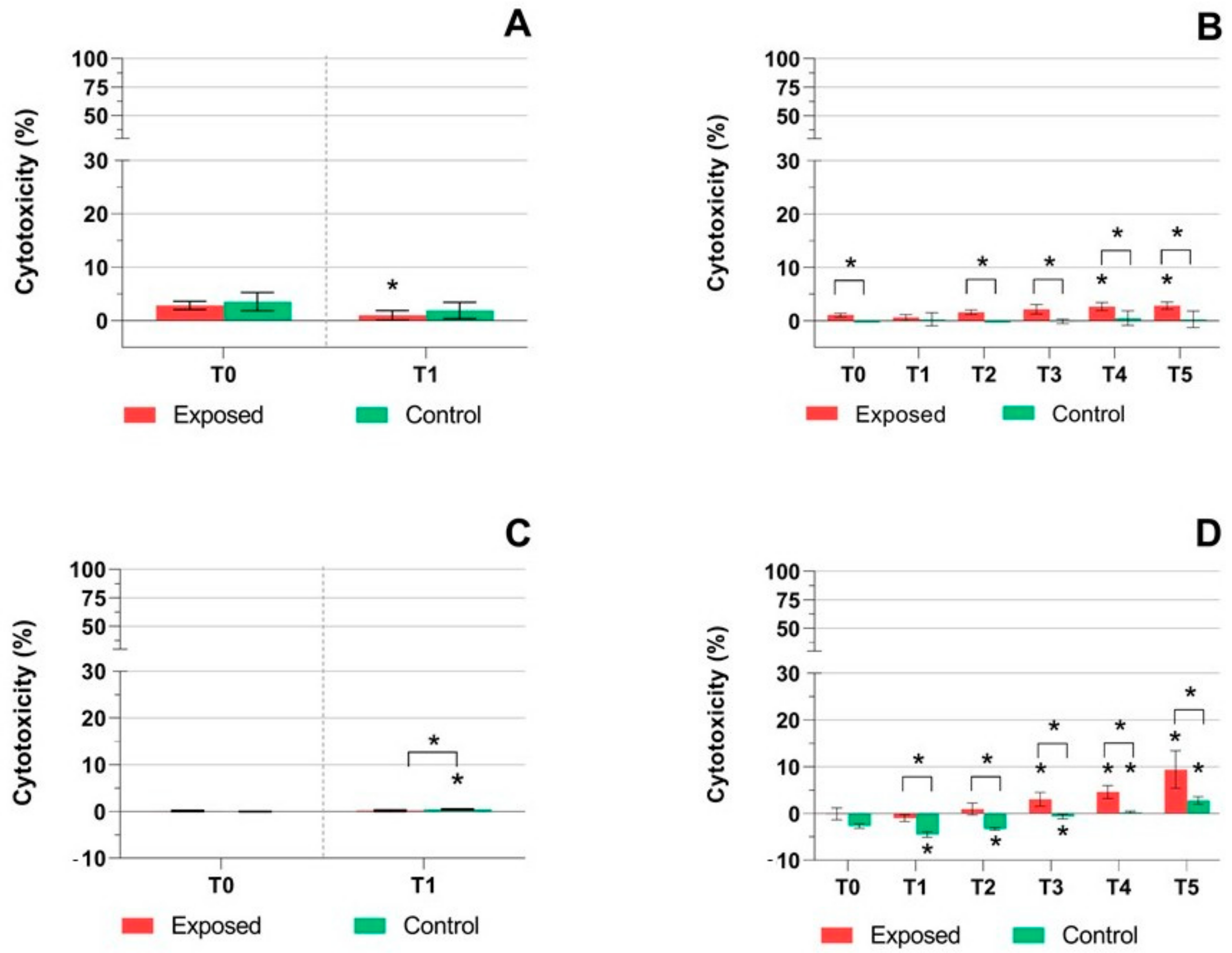
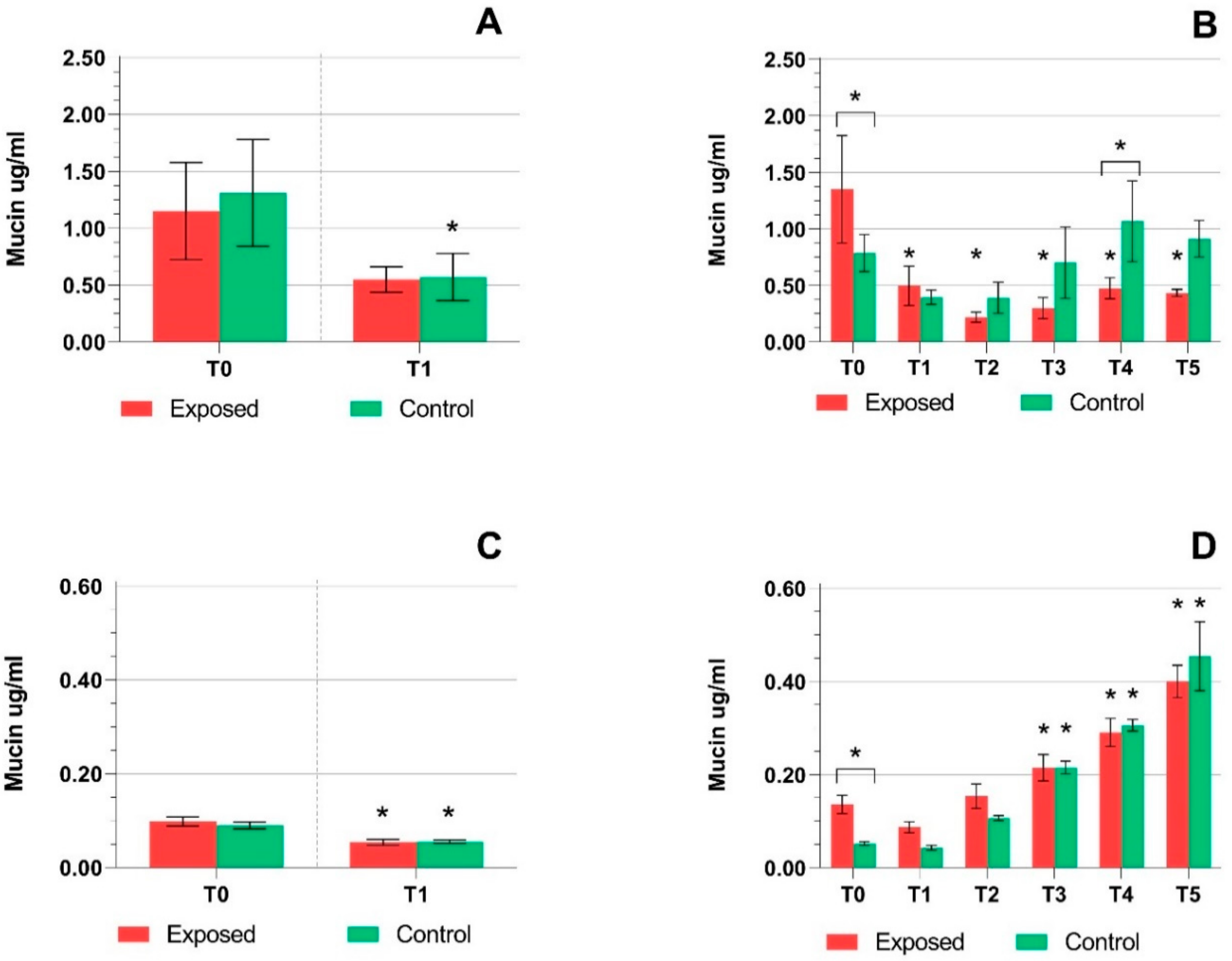
2.1. TEER Measurement, Cytotoxicity Assays, and Mucin Production Quantification
2.2. DNA Breaks Detection
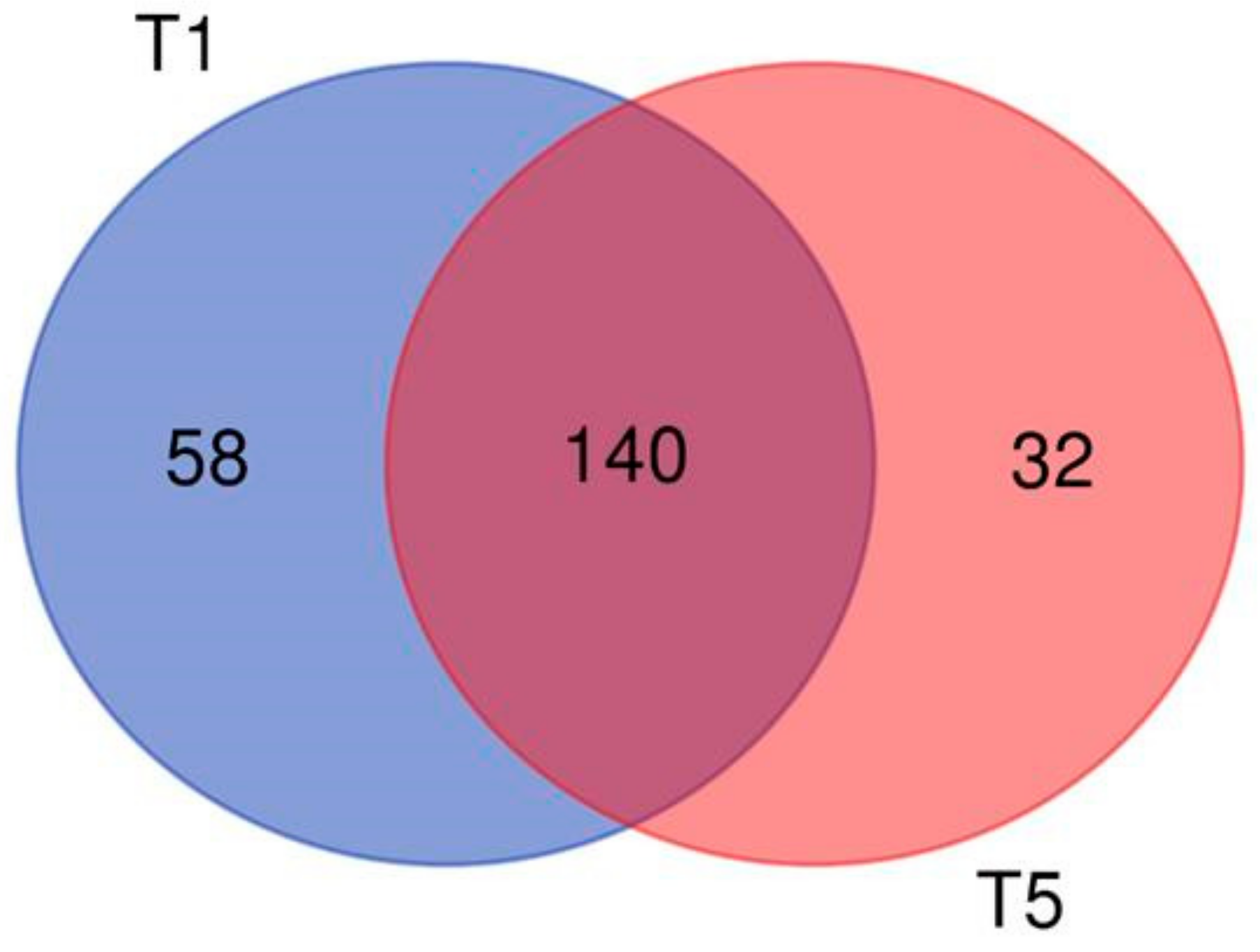
2.3. mRNA Expression Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. The Complete Emissions Exposure System
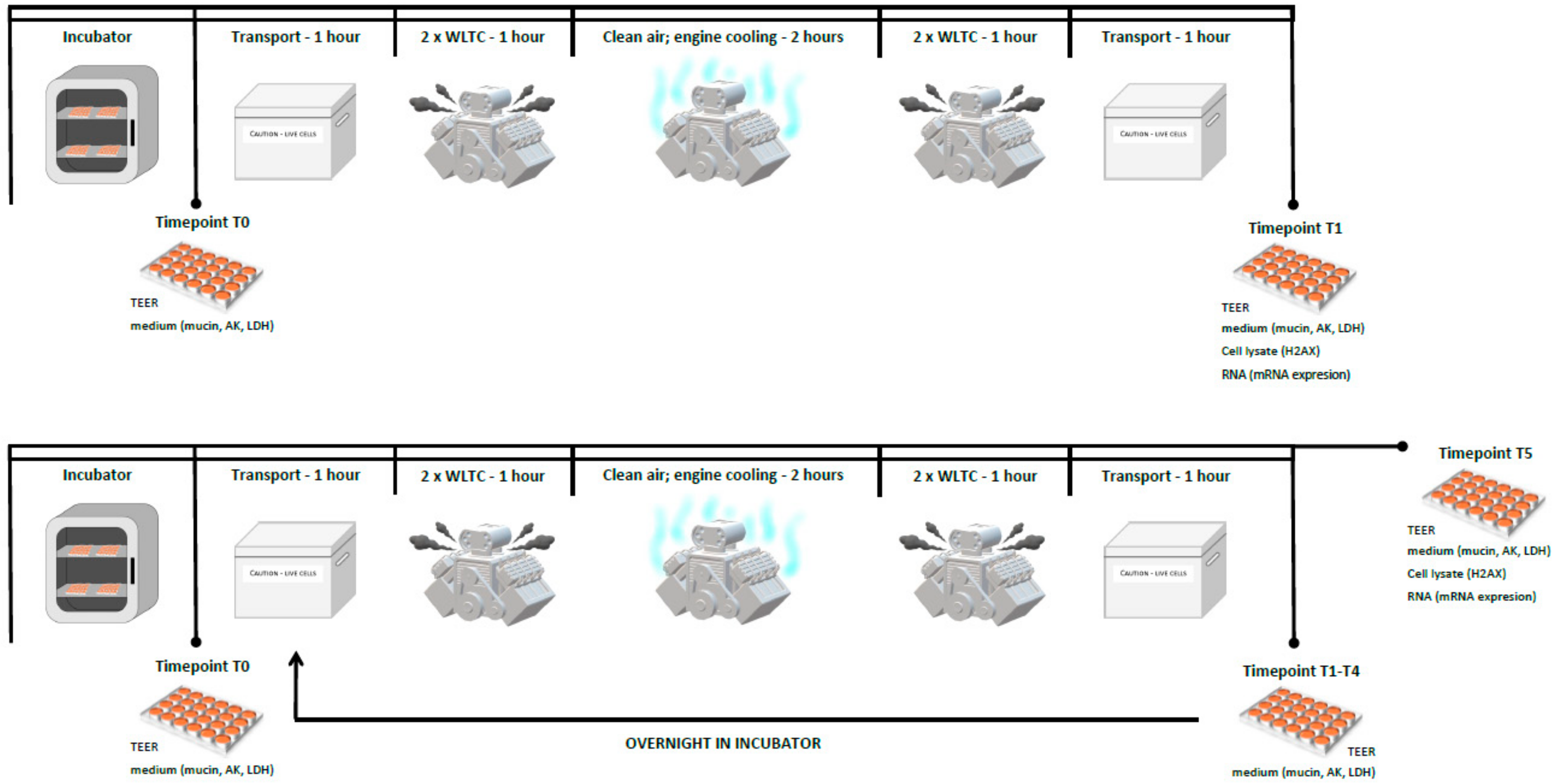
4.3. Exposure Scheme
4.4. TEER Measurement and Mucin Production Quantification
4.5. Cytotoxicity Assays
4.6. Phosphorylation of Histone H2AX
4.7. mRNA Expression Analysis
4.8. Quantitative Real-Time PCR Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AK | Adenylate kinase |
| ALI | Air–liquid interface |
| BEAS-2B | Human bronchial epithelial cells |
| DISI | Direct injection spark ignition engine |
| ELLA | Sandwich enzyme-linked lectin assay |
| EOM | Extractable organic matter |
| ER | Endoplasmic reticulum |
| E5 | Ordinary gasoline, 5% ethanol blend |
| E20 | Gasoline, 20% ethanol blend |
| HPLC | High performance liquid chromatography |
| LDH | Lactate dehydrogenase |
| NOx | Nitrogen oxides |
| PAH | Polycyclic aromatic hydrocarbon |
| PM | Particulate matter |
| qRT-PCR | Quantitative real-time PCR |
| SD | Standard deviation |
| TEER | Transepithelial electrical resistance |
| WLTC | World harmonized light vehicle test cycle |
| ɣ-H2AX | Phosphorylated histone H2AX |
References
- Sram, R.J.; Binkova, B.; Dejmek, J.; Bobak, M. Ambient Air Pollution and Pregnancy Outcomes: A Review of the Literature. Environ. Health Perspect. 2005, 113, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Sram, R.J.; Binkova, B.; Rössner, P.; Rubes, J.; Topinka, J.; Dejmek, J. Adverse Reproductive Outcomes from Exposure to Environmental Mutagens. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1999, 428, 203–215. [Google Scholar] [CrossRef]
- LEWTAS, J. Air Pollution Combustion Emissions: Characterization of Causative Agents and Mechanisms Associated with Cancer, Reproductive, and Cardiovascular Effects. Mutat. Res. Mutat. Res. 2007, 636, 95–133. [Google Scholar] [CrossRef] [PubMed]
- IARC. Diesel Engine Exhaust Carcinogenic; Press Release N° 213; IARC: Lyon, France, 2012. [Google Scholar]
- IARC Monographs. Diesel and Gasoline Engine Exhausts; IARC Monographs: Lyon, France, 2014; Volume 105. [Google Scholar]
- Möhner, M. Driving Ban for Diesel-Powered Vehicles in Major Cities: An Appropriate Penalty for Exceeding the Limit Value for Nitrogen Dioxide? Int. Arch. Occup. Environ. Health 2018, 91, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roth, P.; Ruehl, C.R.; Shafer, M.M.; Antkiewicz, D.S.; Durbin, T.D.; Cocker, D.; Asa-Awuku, A.; Karavalakis, G. Physical, Chemical, and Toxicological Characteristics of Particulate Emissions from Current Technology Gasoline Direct Injection Vehicles. Sci. Total Environ. 2019, 650, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- DeMarini, D.M. Genotoxicity Biomarkers Associated with Exposure to Traffic and Near-Road Atmospheres: A Review. Mutagenesis 2013, 28, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.J.; Castro, N.; Zhang, J.; Rostami, A.; Lucci, F.; Pithawalla, Y.; Kuczaj, A.K.; Gilman, I.G.; Kosachevsky, P.; Hoeng, J.; et al. Deposition Efficiency and Uniformity of Monodisperse Solid Particle Deposition in the Vitrocell® 24/48 Air–Liquid-Interface in Vitro Exposure System. Aerosol Sci. Technol. 2020, 54, 52–65. [Google Scholar] [CrossRef]
- Chan, J.H.; Tsolakis, A.; Herreros, J.M.; Kallis, K.X.; Hergueta, C.; Sittichompoo, S.; Bogarra, M. Combustion, Gaseous Emissions and PM Characteristics of Di-Methyl Carbonate (DMC)-Gasoline Blend on Gasoline Direct Injection (GDI) Engine. Fuel 2020, 263, 116742. [Google Scholar] [CrossRef]
- Yang, J.; Roth, P.; Durbin, T.D.; Shafer, M.M.; Hemming, J.; Antkiewicz, D.S.; Asa-Awuku, A.; Karavalakis, G. Emissions from a Flex Fuel GDI Vehicle Operating on Ethanol Fuels Show Marked Contrasts in Chemical, Physical and Toxicological Characteristics as a Function of Ethanol Content. Sci. Total Environ. 2019, 683, 749–761. [Google Scholar] [CrossRef]
- Carugno, M.; Consonni, D.; Randi, G.; Catelan, D.; Grisotto, L.; Bertazzi, P.A.; Biggeri, A.; Baccini, M. Air Pollution Exposure, Cause-Specific Deaths and Hospitalizations in a Highly Polluted Italian Region. Environ. Res. 2016, 147, 415–424. [Google Scholar] [CrossRef]
- Luyts, K.; Napierska, D.; Dinsdale, D.; Klein, S.G.; Serchi, T.; Hoet, P.H.M. A Coculture Model of the Lung-Blood Barrier: The Role of Activated Phagocytic Cells. Toxicol. Vitr. 2014, 29, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.K.; de Mallia, J.O.; Singh, N.; Chapman, K.E.; Doak, S.H.; Jenkins, G.J.S. A Three-Dimensional in Vitro HepG2 Cells Liver Spheroid Model for Genotoxicity Studies. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 825, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Furubayashi, T.; Inoue, D.; Nishiyama, N.; Tanaka, A.; Yutani, R.; Kimura, S.; Katsumi, H.; Yamamoto, A.; Sakane, T. Comparison of Various Cell Lines and Three-Dimensional Mucociliary Tissue Model Systems to Estimate Drug Permeability Using an In Vitro Transport Study to Predict Nasal Drug Absorption in Rats. Pharmaceutics 2020, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Ridley, C.; Thornton, D.J. Mucins: The Frontline Defence of the Lung. Biochem. Soc. Trans. 2018, 46, 1099–1106. [Google Scholar] [CrossRef]
- Tratnjek, L.; Kreft, M.; Kristan, K.; Kreft, M.E. Ciliary Beat Frequency of in Vitro Human Nasal Epithelium Measured with the Simple High-Speed Microscopy Is Applicable for Safety Studies of Nasal Drug Formulations. Toxicol. Vitr. 2020, 66, 104865. [Google Scholar] [CrossRef]
- Loiseau, E.; Gsell, S.; Nommick, A.; Jomard, C.; Gras, D.; Chanez, P.; D’Ortona, U.; Kodjabachian, L.; Favier, J.; Viallat, A. Active Mucus–Cilia Hydrodynamic Coupling Drives Self-Organization of Human Bronchial Epithelium. Nat. Phys. 2020. [Google Scholar] [CrossRef]
- Arora, P.; Sharma, S.; Garg, S. Permeability Issues in Nasal Drug Delivery. Drug Discov. Today 2002, 7, 967–975. [Google Scholar] [CrossRef]
- Mercier, C.; Jacqueroux, E.; He, Z.; Hodin, S.; Constant, S.; Perek, N.; Boudard, D.; Delavenne, X. Pharmacological Characterization of the 3D MucilAirTM Nasal Model. Eur. J. Pharm. Biopharm. 2019, 139, 186–196. [Google Scholar] [CrossRef]
- Kletting, S.; Barthold, S.; Repnik, U.; Griffiths, G.; Loretz, B.; Schneider-Daum, N.; de Carvalho-Wodarz, C.S.; Lehr, C.-M.M. Co-Culture of Human Alveolar Epithelial (HAELVi) and Macrophage (THP-1) Cell Lines. ALTEX 2018, 35, 211–222. [Google Scholar] [CrossRef]
- Huang, S.; Wiszniewski, L.; Constant, S.; Roggen, E. Potential of in Vitro Reconstituted 3D Human Airway Epithelia (MucilAirTM) to Assess Respiratory Sensitizers. Toxicol. Vitr. 2013, 27, 1151–1156. [Google Scholar] [CrossRef]
- Huang, S.; Boda, B.; Vernaz, J.; Ferreira, E.; Wiszniewski, L.; Constant, S. Establishment and Characterization of an in Vitro Human Small Airway Model (SmallAir TM ). Eur. J. Pharm. Biopharm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Balharry, D.; Sexton, K.; Bérubé, K.A. An in Vitro Approach to Assess the Toxicity of Inhaled Tobacco Smoke Components: Nicotine, Cadmium, Formaldehyde and Urethane. Toxicology 2008, 244, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Comte, P.; Czerwinski, J.; Kasper, M.; Mayer, A.C.R.; Gehr, P.; Burtscher, H.; Morin, J.-P.; Konstandopoulos, A.; Rothen-Rutishauser, B. New Exposure System To Evaluate the Toxicity of (Scooter) Exhaust Emissions in Lung Cells in Vitro. Environ. Sci. Technol. 2010, 44, 2632–2638. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Mueller, L.; Popovicheva, O.B.; Raemy, D.O.; Czerwinski, J.; Comte, P.; Mayer, A.; Gehr, P.; Rothen-Rutishauser, B.; Clift, M.J.D. Cerium Dioxide Nanoparticles Can Interfere with the Associated Cellular Mechanistic Response to Diesel Exhaust Exposure. Toxicol. Lett. 2012, 214, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hawley, B.; L’Orange, C.; Olsen, D.B.; Marchese, A.J.; Volckens, J. Oxidative Stress and Aromatic Hydrocarbon Response of Human Bronchial Epithelial Cells Exposed to Petro- or Biodiesel Exhaust Treated with a Diesel Particulate Filter. Toxicol. Sci. 2014, 141, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Jonsdottir, H.R.; Delaval, M.; Leni, Z.; Keller, A.; Brem, B.T.; Siegerist, F.; Schönenberger, D.; Durdina, L.; Elser, M.; Burtscher, H.; et al. Non-Volatile Particle Emissions from Aircraft Turbine Engines at Ground-Idle Induce Oxidative Stress in Bronchial Cells. Commun. Biol. 2019, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Dailey, L.A.; Soukup, J.M.; Stonehuerner, J.; Richards, J.H.; Devlin, R.B. Growth of Human Bronchial Epithelial Cells at an Air-Liquid Interface Alters the Response to Particle Exposure. Part. Fibre Toxicol. 2013, 10, 1. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Låg, M.; Lilleaas, E.; Cassee, F.; Schwarze, P. Differential Proinflammatory Responses Induced by Diesel Exhaust Particles with Contrasting PAH and Metal Content. Environ. Toxicol. 2015, 30, 188–196. [Google Scholar] [CrossRef]
- Goudarzi, G.; Shirmardi, M.; Naimabadi, A.; Ghadiri, A.; Sajedifar, J. Chemical and Organic Characteristics of PM2.5 Particles and Their in-Vitro Cytotoxic Effects on Lung Cells: The Middle East Dust Storms in Ahvaz, Iran. Sci. Total Environ. 2019, 655, 434–445. [Google Scholar] [CrossRef]
- Libalova, H.; Rossner, P.; Vrbova, K.; Brzicova, T.; Sikorova, J.; Vojtisek-Lom, M.; Beranek, V.; Klema, J.; Ciganek, M.; Neca, J.; et al. Transcriptional Response to Organic Compounds from Diverse Gasoline and Biogasoline Fuel Emissions in Human Lung Cells. Toxicol. Vitr. 2018, 48, 329–341. [Google Scholar] [CrossRef]
- Velali, E.; Papachristou, E.; Pantazaki, A.; Choli-Papadopoulou, T.; Argyrou, N.; Tsourouktsoglou, T.; Lialiaris, S.; Constantinidis, A.; Lykidis, D.; Lialiaris, T.S.; et al. Cytotoxicity and Genotoxicity Induced in Vitro by Solvent-Extractable Organic Matter of Size-Segregated Urban Particulate Matter. Environ. Pollut. 2016, 218, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Velali, E.; Papachristou, E.; Pantazaki, A.; Choli-Papadopoulou, T.; Planou, S.; Kouras, A.; Manoli, E.; Besis, A.; Voutsa, D.; Samara, C. Redox Activity and in Vitro Bioactivity of the Water-Soluble Fraction of Urban Particulate Matter in Relation to Particle Size and Chemical Composition. Environ. Pollut. 2016, 208, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Vojtisek-Lom, M.; Pechout, M.; Macoun, D.; Rameswaran, R.; Praharaj, K.K.; Cervena, T.; Topinka, J.; Rossner, P. Assessing Exhaust Toxicity with Biological Detector: Configuration of Portable Air-Liquid Interface Human Lung Cell Model Exposure System, Sampling Train and Test Conditions. SAE Int. J. Adv. Curr. Pract. Mobil. 2019. [Google Scholar] [CrossRef]
- Steiner, S.; Czerwinski, J.; Comte, P.; Heeb, N.V.; Mayer, A.; Petri-Fink, A.; Rothen-Rutishauser, B. Effects of an Iron-Based Fuel-Borne Catalyst and a Diesel Particle Filter on Exhaust Toxicity in Lung Cells in Vitro. Anal. Bioanal. Chem. 2015, 407, 5977–5986. [Google Scholar] [CrossRef]
- Bisig, C.; Comte, P.; Güdel, M.; Czerwinski, J.; Mayer, A.; Müller, L.; Petri-Fink, A.; Rothen-Rutishauser, B. Assessment of Lung Cell Toxicity of Various Gasoline Engine Exhausts Using a Versatile in Vitro Exposure System. Environ. Pollut. 2018, 235, 263–271. [Google Scholar] [CrossRef]
- Bisig, C.; Roth, M.; Müller, L.; Comte, P.; Heeb, N.; Mayer, A.; Czerwinski, J.; Petri-Fink, A.; Rothen-Rutishauser, B. Hazard Identification of Exhausts from Gasoline-Ethanol Fuel Blends Using a Multi-Cellular Human Lung Model. Environ. Res. 2016, 151, 789–796. [Google Scholar] [CrossRef]
- Aufderheide, M.; Scheffler, S.; Ito, S.; Ishikawa, S.; Emura, M. Ciliatoxicity in Human Primary Bronchiolar Epithelial Cells after Repeated Exposure at the Air–Liquid Interface with Native Mainstream Smoke of K3R4F Cigarettes with and without Charcoal Filter. Exp. Toxicol. Pathol. 2015, 67, 407–411. [Google Scholar] [CrossRef]
- Chortarea, S.; Clift, M.J.D.; Vanhecke, D.; Endes, C.; Wick, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Repeated Exposure to Carbon Nanotube-Based Aerosols Does Not Affect the Functional Properties of a 3D Human Epithelial Airway Model. Nanotoxicology 2015, 5390, 1–11. [Google Scholar] [CrossRef]
- Méausoone, C.; El Khawaja, R.; Tremolet, G.; Siffert, S.; Cousin, R.; Cazier, F.; Billet, S.; Courcot, D.; Landkocz, Y. In Vitro Toxicological Evaluation of Emissions from Catalytic Oxidation Removal of Industrial VOCs by Air/Liquid Interface (ALI) Exposure System in Repeated Mode. Toxicol. Vitr. 2019, 58, 110–117. [Google Scholar] [CrossRef]
- Ito, S.; Matsumura, K.; Ishimori, K.; Ishikawa, S. In Vitro Long-term Repeated Exposure and Exposure Switching of a Novel Tobacco Vapor Product in a Human Organotypic Culture of Bronchial Epithelial Cells. J. Appl. Toxicol. 2020, 40, 1248–1258. [Google Scholar] [CrossRef]
- Di Cristo, L.; Grimaldi, B.; Catelani, T.; Vázquez, E.; Pompa, P.P.; Sabella, S. Repeated Exposure to Aerosolized Graphene Oxide Mediates Autophagy Inhibition and Inflammation in a Three-Dimensional Human Airway Model. Mater. Today Bio 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Matsumura, K.; Kitamura, N.; Takanami, Y.; Ito, S. Multi-Omics Analysis: Repeated Exposure of a 3D Bronchial Tissue Culture to Whole-Cigarette Smoke. Toxicol. Vitr. 2019, 54, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Svecova, V.; Topinka, J.; Solansky, I.; Rossner, P.; Sram, R.J. Personal Exposure to Carcinogenic Polycyclic Aromatic Hydrocarbons in the Czech Republic. J. Expo. Sci. Environ. Epidemiol. 2013, 23, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Good, N.; Mölter, A.; Ackerson, C.; Bachand, A.; Carpenter, T.; Clark, M.L.; Fedak, K.M.; Kayne, A.; Koehler, K.; Moore, B.; et al. The Fort Collins Commuter Study: Impact of Route Type and Transport Mode on Personal Exposure to Multiple Air Pollutants. J. Expo. Sci. Environ. Epidemiol. 2016, 26, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Campen, M.; Robertson, S.; Lund, A.; Lucero, J.; McDonald, J. Engine Exhaust Particulate and Gas Phase Contributions to Vascular Toxicity. Inhal. Toxicol. 2014, 26, 353–360. [Google Scholar] [CrossRef]
- Kooter, I.M.; Alblas, M.J.; Jedynska, A.D.; Steenhof, M.; Houtzager, M.M.G.; Van Ras, M. Alveolar Epithelial Cells (A549) Exposed at the Air–Liquid Interface to Diesel Exhaust: First Study in TNO’s Powertrain Test Center. Toxicol. Vitr. 2013, 27, 2342–2349. [Google Scholar] [CrossRef]
- Cao, X.; Muskhelishvili, L.; Latendresse, J.; Richter, P.; Heflich, R.H. Evaluating the Toxicity of Cigarette Whole Smoke Solutions in an Air-Liquid-Interface Human in Vitro Airway Tissue Model. Toxicol. Sci. 2017, 1156, 14–24. [Google Scholar] [CrossRef]
- Reddel, R.R.; Ke, Y.; Gerwin, B.I.; McMenamin, M.G.; Lechner, J.F.; Su, R.T.; Brash, D.E.; Park, J.B.; Rhim, J.S.; Harris, C.C. Transformation of Human Bronchial Epithelial Cells by Infection with SV40 or Adenovirus-12 SV40 Hybrid Virus, or Transfection via Strontium Phosphate Coprecipitation with a Plasmid Containing SV40 Early Region Genes. Cancer Res. 1988, 48, 1904–1909. [Google Scholar]
- Rossner, P.; Cervena, T.; Vojtisek-Lom, M.; Vrbova, K.; Ambroz, A.; Novakova, Z.; Elzeinova, F.; Margaryan, H.; Beranek, V.; Pechout, M.; et al. The Biological Effects of Complete Gasoline Engine Emissions Exposure in a 3D Human Airway Model (MucilAirTM) and in Human Bronchial Epithelial Cells (BEAS-2B). Int. J. Mol. Sci. 2019, 20, 5710. [Google Scholar] [CrossRef]
- Benson, K.; Cramer, S.; Galla, H.J. Impedance-Based Cell Monitoring: Barrier Properties and Beyond. Fluids Barriers CNS 2013, 10, 1–11. [Google Scholar] [CrossRef]
- Nalayanda, D.D.; Fulton, W.B.; Colombani, P.M.; Wang, T.H.; Abdullah, F. Pressure Induced Lung Injury in a Novel in Vitro Model of the Alveolar Interface: Protective Effect of Dexamethasone. J. Pediatr. Surg. 2014, 49, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate Matter 2.5 Damages Skin Cells by Inducing Oxidative Stress, Subcellular Organelle Dysfunction, and Apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Watterson, T.L.; Hamilton, B.; Martin, R.; Coulombe, R.A. Urban Particulate Matter Causes ER Stress and the Unfolded Protein Response in Human Lung Cells. Toxicol. Sci. 2009, 112, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Trieu, D.; Waddell, T.K.; McGuigan, A.P. A Microfluidic Device to Apply Shear Stresses to Polarizing Ciliated Airway Epithelium Using Air Flow. Biomicrofluidics 2014, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Canton, C.; Anadón, A.; Meredith, C. ΓH2AX as a Novel Endpoint to Detect DNA Damage: Applications for the Assessment of the in Vitro Genotoxicity of Cigarette Smoke. Toxicol. Vitr. 2012, 26, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Riu, A.; Jacques, C.; Hillenweck, A.; Jamin, E.L.; Zalko, D.; Cravedi, J.P. Use of the ΓH2AX Assay for Assessing the Genotoxicity of Polycyclic Aromatic Hydrocarbons in Human Cell Lines. Toxicol. Lett. 2010, 199, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Oya, E.; Ovrevik, J.; Arlt, V.M.; Nagy, E.; Phillips, D.H.; Holme, J.A. DNA Damage and DNA Damage Response in Human Bronchial Epithelial BEAS-2B Cells Following Exposure to 2-Nitrobenzanthrone and 3-Nitrobenzanthrone: Role in Apoptosis. Mutagenesis 2011, 26, 697–708. [Google Scholar] [CrossRef]
- Rossner, P.; Rossnerova, A.; Beskid, O.; Tabashidze, N.; Libalova, H.; Uhlirova, K.; Topinka, J.; Sram, R.J. Nonhomologous DNA End Joining and Chromosome Aberrations in Human Embryonic Lung Fibroblasts Treated with Environmental Pollutants. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2014, 763–764, 28–38. [Google Scholar] [CrossRef]
- Toyooka, T.; Shinmen, T.; Aarts, J.M.M.J.G.; Ibuki, Y. Dual Effects of N-Acetyl-l-Cysteine Dependent on NQO1 Activity: Suppressive or Promotive of 9,10-Phenanthrenequinone-Induced Toxicity. Toxicol. Appl. Pharmacol. 2012, 264, 404–412. [Google Scholar] [CrossRef]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER Stress Suppresses DNA Double-Strand Break Repair and Sensitizes Tumor Cells to Ionizing Radiation by Stimulating Proteasomal Degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef]
- Tomašek, I.; Horwell, C.J.; Bisig, C.; Damby, D.E.; Comte, P.; Czerwinski, J.; Petri-Fink, A.; Clift, M.J.D.; Drasler, B.; Rothen-Rutishauser, B. Respiratory Hazard Assessment of Combined Exposure to Complete Gasoline Exhaust and Respirable Volcanic Ash in a Multicellular Human Lung Model at the Air-Liquid Interface. Environ. Pollut. 2018, 238, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Cervena, T.; Vrbova, K.; Rossnerova, A.; Topinka, J.; Rossner, P. Short-Term and Long-Term Exposure of the MucilAirTM Model to Polycyclic Aromatic Hydrocarbons. Altern. Lab. Anim. 2019, 47, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Rabson, A.B.; Germino, J.F.; Gallo, M.A.; Tian, Y. Mechanism of Suppression of Cytochrome P-450 1A1 Expression by Tumor Necrosis Factor-α and Lipopolysaccharide. J. Biol. Chem. 2001, 276, 39638–39644. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Koike, E.; Kobayashi, T. Microarray Analysis of Gene Expression in Rat Alveolar Epithelial Cells Exposed to Fractionated Organic Extracts of Diesel Exhaust Particles. Toxicology 2009, 262, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Peltzer, A.; Fillinger, S.; Alneberg, J.; Patel, H.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. Nf-Core: Community Curated Bioinformatics Pipelines. bioRxiv 2019, 610741. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research 2009, 37, W305–W311. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure Time | Gene Name | Ensemble ID | Biological Pathway | Log2 FC | Adj. p-Value |
|---|---|---|---|---|---|
| Five days | CYP1A1 | ENSG00000140465 | Cytochrome P450s and Phase I Drug Metabolism | −2.78 | 0.019 |
| GSTA3 | ENSG00000174156 | Immunotoxicity | 1.46 | 0.019 |
| A. The expression of genes induced by exposure to complete emissions in BEAS-2B cells for one day | |||||
| Exposure Time | Gene Name | Ensemble ID | Biological Pathway | Log2 FC | Adj. p-Value |
| One day | BID | ENSG00000015475 | Apoptosis | 1.64 | 0.015 |
| CASP1 | ENSG00000137752 | Apoptosis | 1.33 | 0.023 | |
| TNFSF10 | ENSG00000121858 | Apoptosis | 0.96 | 0.047 | |
| ESD | ENSG00000139684 | Cytochrome P450s and Phase I Drug Metabolism | 1.93 | 0.008 | |
| MAOB | ENSG00000069535 | Cytochrome P450s and Phase I Drug Metabolism | −0.84 | 0.043 | |
| MLH1 | ENSG00000076242 | DNA Damage and Repair | 2.41 | 0.006 | |
| MSH2 | ENSG00000095002 | DNA Damage and Repair | 2.09 | 0.008 | |
| PARP1 | ENSG00000143799 | DNA Damage and Repair | 1.54 | 0.014 | |
| PCNA | ENSG00000132646 | DNA Damage and Repair | 1.89 | 0.027 | |
| ATF4 | ENSG00000128272 | Endoplasmic Reticulum Stress and Unfolded Protein Response | 1.52 | 0.023 | |
| DERL1 | ENSG00000136986 | Endoplasmic Reticulum Stress and Unfolded Protein Response | 1.87 | 0.013 | |
| HERPUD1 | ENSG00000051108 | Endoplasmic Reticulum Stress and Unfolded Protein Response | 2.05 | 0.007 | |
| SERP1 | ENSG00000120742 | Endoplasmic Reticulum Stress and Unfolded Protein Response | 1.37 | 0.019 | |
| SYVN1 | ENSG00000162298 | Endoplasmic Reticulum Stress and Unfolded Protein Response | 1.91 | 0.014 | |
| ACAA2 | ENSG00000167315 | Fatty Acid Metabolism | 2.54 | 0.006 | |
| BDH2 | ENSG00000164039 | Fatty Acid Metabolism | 1.55 | 0.019 | |
| DNAJA1 | ENSG00000086061 | Heat Shock Response | 1.60 | 0.014 | |
| DNAJA3 | ENSG00000103423 | Heat Shock Response | 2.10 | 0.008 | |
| HSP90B1 | ENSG00000166598 | Heat Shock Response | 2.44 | 0.008 | |
| HSPA1A | ENSG00000204389 | Heat Shock Response | 2.01 | 0.008 | |
| HSPA8 | ENSG00000109971 | Heat Shock Response | 1.96 | 0.008 | |
| HSPA9 | ENSG00000113013 | Heat Shock Response | 1.29 | 0.027 | |
| HSPD1 | ENSG00000144381 | Heat Shock Response | 1.55 | 0.014 | |
| HSPE1 | ENSG00000115541 | Heat Shock Response | 2.72 | 0.006 | |
| TCP1 | ENSG00000120438 | Heat Shock Response | 2.05 | 0.008 | |
| METAP2 | ENSG00000111142 | Immunotoxicity | 1.26 | 0.019 | |
| MDH1 | ENSG00000014641 | Mitochondrial Energy Metabolism | 1.97 | 0.008 | |
| MDH2 | ENSG00000146701 | Mitochondrial Energy Metabolism | 1.92 | 0.014 | |
| SDHD | ENSG00000204370 | Mitochondrial Energy Metabolism | 1.90 | 0.008 | |
| SUCLA2 | ENSG00000136143 | Mitochondrial Energy Metabolism | 1.45 | 0.020 | |
| HOXA3 | ENSG00000105997 | Necrosis | 2.24 | 0.008 | |
| GPX1 | ENSG00000233276 | Oxidative Stress and Antioxidant Response | 1.47 | 0.027 | |
| SOD1 | ENSG00000142168 | Oxidative Stress and Antioxidant Response | 2.16 | 0.008 | |
| ASAH1 | ENSG00000104763 | Phospholipidosis | 2.36 | 0.006 | |
| MRPS18B | ENSG00000204568 | Phospholipidosis | 1.93 | 0.010 | |
| ADK | ENSG00000156110 | Steatosis | 1.33 | 0.019 | |
| COMT | ENSG00000093010 | Steatosis | 2.08 | 0.008 | |
| HADHB | ENSG00000138029 | Steatosis | 2.47 | 0.006 | |
| LY6D | ENSG00000167656 | Steatosis | 2.41 | 0.008 | |
| B. The expression of genes induced by exposure to complete emissions in BEAS-2B cells for five days | |||||
| Exposure Time | Gene Name | Ensemble ID | Biological Pathway | Log2 FC | Adj. p-Value |
| Five day | BIRC3 | ENSG00000023445 | Apoptosis | −0.971 | 0.000 |
| MCL1 | ENSG00000143384 | Apoptosis | −0.285 | 0.038 | |
| IL6 | ENSG00000136244 | Cholestasis and Immunotoxicity | −2.227 | 0.000 | |
| MDM2 | ENSG00000135679 | DNA Damage and Repair | 0.548 | 0.006 | |
| ACOX1 | ENSG00000161533 | Fatty Acid Metabolism | 0.575 | 0.024 | |
| CPT2 | ENSG00000157184 | Fatty Acid Metabolism | 0.472 | 0.038 | |
| IL1A | ENSG00000115008 | Immunotoxicity | −1.118 | 0.000 | |
| PTGS2 | ENSG00000073756 | Immunotoxicity | −1.002 | 0.018 | |
| IDH3B | ENSG00000101365 | Mitochondrial Energy Metabolism | −0.480 | 0.018 | |
| NUDT15 | ENSG00000136159 | Oxidative Stress and Antioxidant Response | −0.557 | 0.018 | |
| SLC2A3 | ENSG00000059804 | Phospholipidosis | −0.989 | 0.028 | |
| Pathway ID | Name | q-Value | Deregulated Genes (N) | Genes in Pathway (N) |
|---|---|---|---|---|
| BEAS-2B, 1-day exposure | ||||
| 413348 | Citrate cycle | <0.001 | 4 | 14 |
| 83045 | Mismatch repair | <0.001 | 3 | 23 |
| BEAS-2B, 5-day exposure | ||||
| 812256 | TNF signaling pathway | <0.01 | 3 | 108 |
| 83105 | Pathways in cancer | <0.01 | 4 | 395 |
| MucilAir™ vs. BEAS-2B, 1-day exposure | ||||
| 868084 | Fatty acid metabolism | <0.001 | 8 | 48 |
| 814926 | Carbon metabolism | <0.001 | 8 | 114 |
| MucilAir™ vs. BEAS-2B, 5-day exposure | ||||
| 83060 | Apoptosis | <0.001 | 6 | 138 |
| 413381 | beta-Oxidation | <0.001 | 3 | 12 |
| 83055 | p53 signaling pathway | 0.001 | 4 | 69 |
| MucilAir™ vs. BEAS-2B, regardless of the exposure period | ||||
| 83060 | Apoptosis | <0.001 | 16 | 138 |
| 82927 | Citrate cycle | <0.001 | 9 | 30 |
| 83032 | Drug metabolism—cytochrome P450 | <0.001 | 11 | 70 |
| Exhaust | WLTC | Average Filter Loading Per Test [mg] | PM Mean [mg/m3] | Average Particle Number Concentrations [#/cm3] |
|---|---|---|---|---|
| Diluted 10:1 exhaust to cells | Cold start | 0.438 ± 0.174 | 0.201 ± 0.089 | 5–8 × 105 |
| Warm start | 0.333 ± 0.099 | 0.148 ± 0.050 | 2 × 105 | |
| Average | 0.385 | 0.175 | ||
| From filter measurements | From size distributions (EEPS, with dilution at 150 °C) | From size distributions (ELPI, no dilution at 160 °C) | From photoacoustic measurements (soot) | |
| Mass concentrations in undiluted exhaust [mg/m3] | 1.75 | ~0.6–0.7 | ~0.6–0.7 | ~0.3 |
| Exposure | Flow Rate Per Insert [cm3/min] | Total Volume of Diluted Exhaust Per Insert [dm3] | Total PM in Diluted Exhaust Per Insert [ng] | Deposition Per Insert (Estimated Deposition Rate 2%) [ng] |
|---|---|---|---|---|
| One-day | 25 | 3 | 525 | ≈10 |
| Five-day | 25 | 15 | 2625 | ≈50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervena, T.; Vojtisek-Lom, M.; Vrbova, K.; Ambroz, A.; Novakova, Z.; Elzeinova, F.; Sima, M.; Beranek, V.; Pechout, M.; Macoun, D.; et al. Ordinary Gasoline Emissions Induce a Toxic Response in Bronchial Cells Grown at Air-Liquid Interface. Int. J. Mol. Sci. 2021, 22, 79. https://doi.org/10.3390/ijms22010079
Cervena T, Vojtisek-Lom M, Vrbova K, Ambroz A, Novakova Z, Elzeinova F, Sima M, Beranek V, Pechout M, Macoun D, et al. Ordinary Gasoline Emissions Induce a Toxic Response in Bronchial Cells Grown at Air-Liquid Interface. International Journal of Molecular Sciences. 2021; 22(1):79. https://doi.org/10.3390/ijms22010079
Chicago/Turabian StyleCervena, Tereza, Michal Vojtisek-Lom, Kristyna Vrbova, Antonin Ambroz, Zuzana Novakova, Fatima Elzeinova, Michal Sima, Vit Beranek, Martin Pechout, David Macoun, and et al. 2021. "Ordinary Gasoline Emissions Induce a Toxic Response in Bronchial Cells Grown at Air-Liquid Interface" International Journal of Molecular Sciences 22, no. 1: 79. https://doi.org/10.3390/ijms22010079
APA StyleCervena, T., Vojtisek-Lom, M., Vrbova, K., Ambroz, A., Novakova, Z., Elzeinova, F., Sima, M., Beranek, V., Pechout, M., Macoun, D., Klema, J., Rossnerova, A., Ciganek, M., Topinka, J., & Rossner, P., Jr. (2021). Ordinary Gasoline Emissions Induce a Toxic Response in Bronchial Cells Grown at Air-Liquid Interface. International Journal of Molecular Sciences, 22(1), 79. https://doi.org/10.3390/ijms22010079