Tracking of Glycans Structure and Metallomics Profiles in BRAF Mutated Melanoma Cells Treated with Vemurafenib


 ,
,  ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
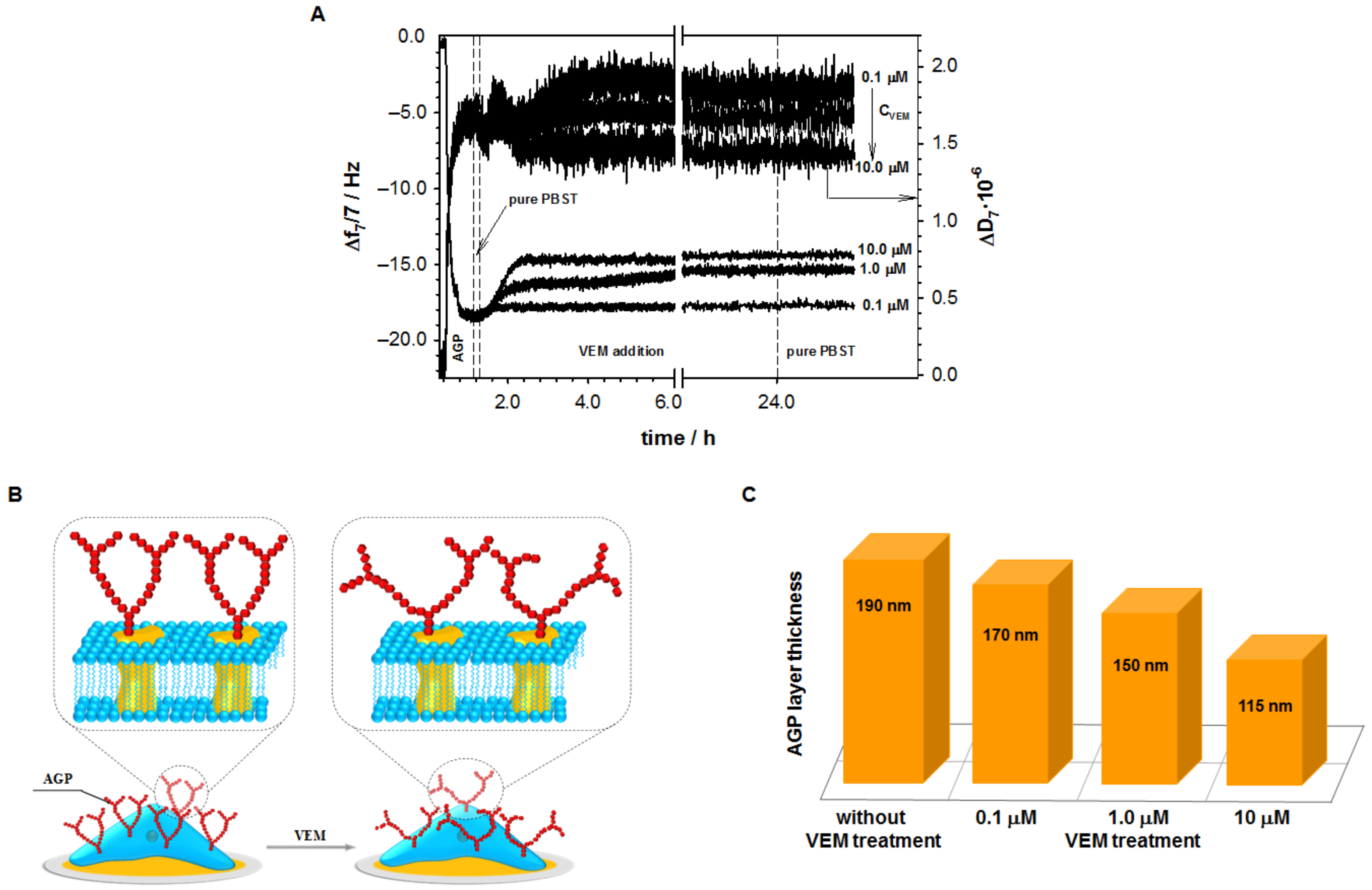
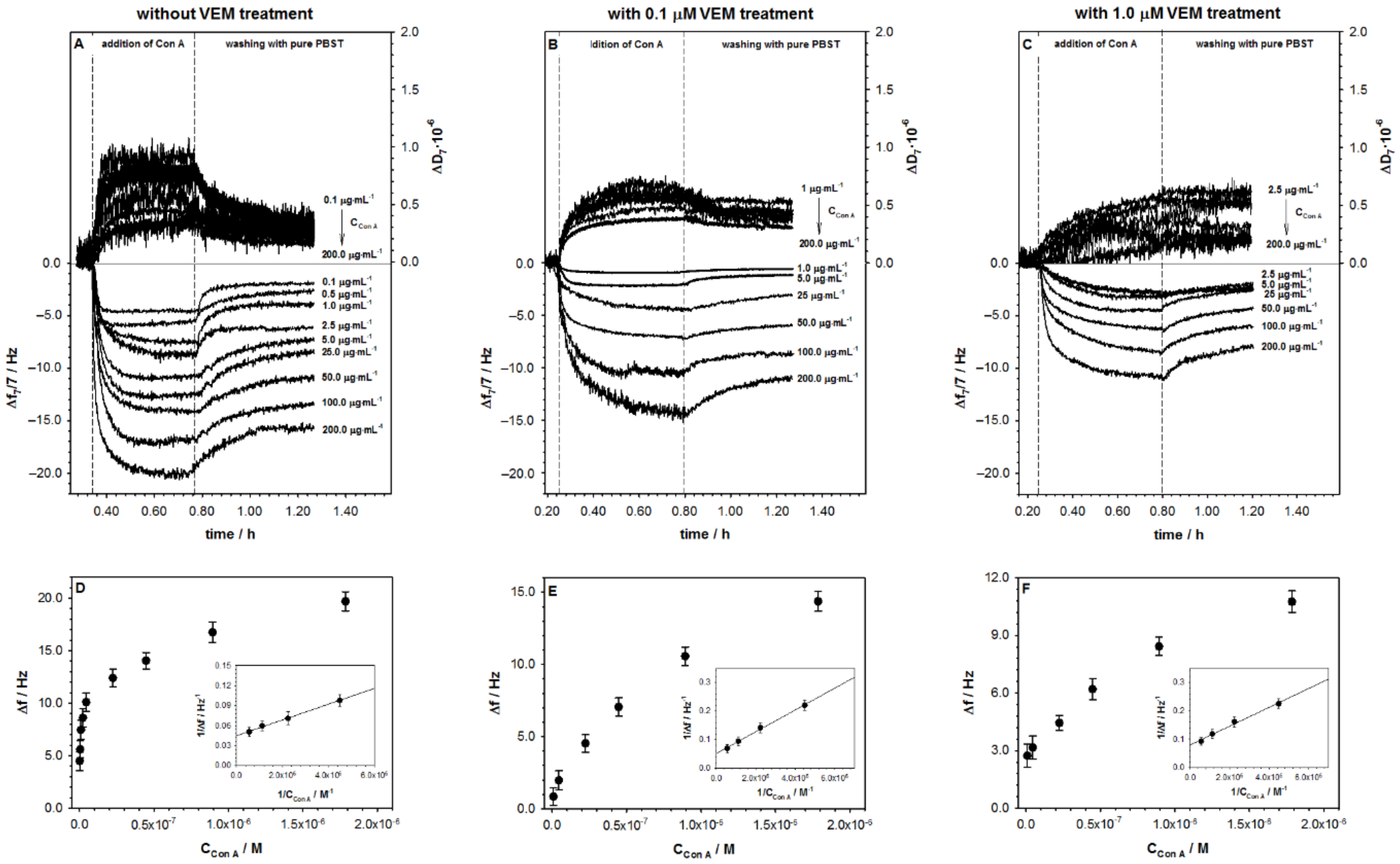
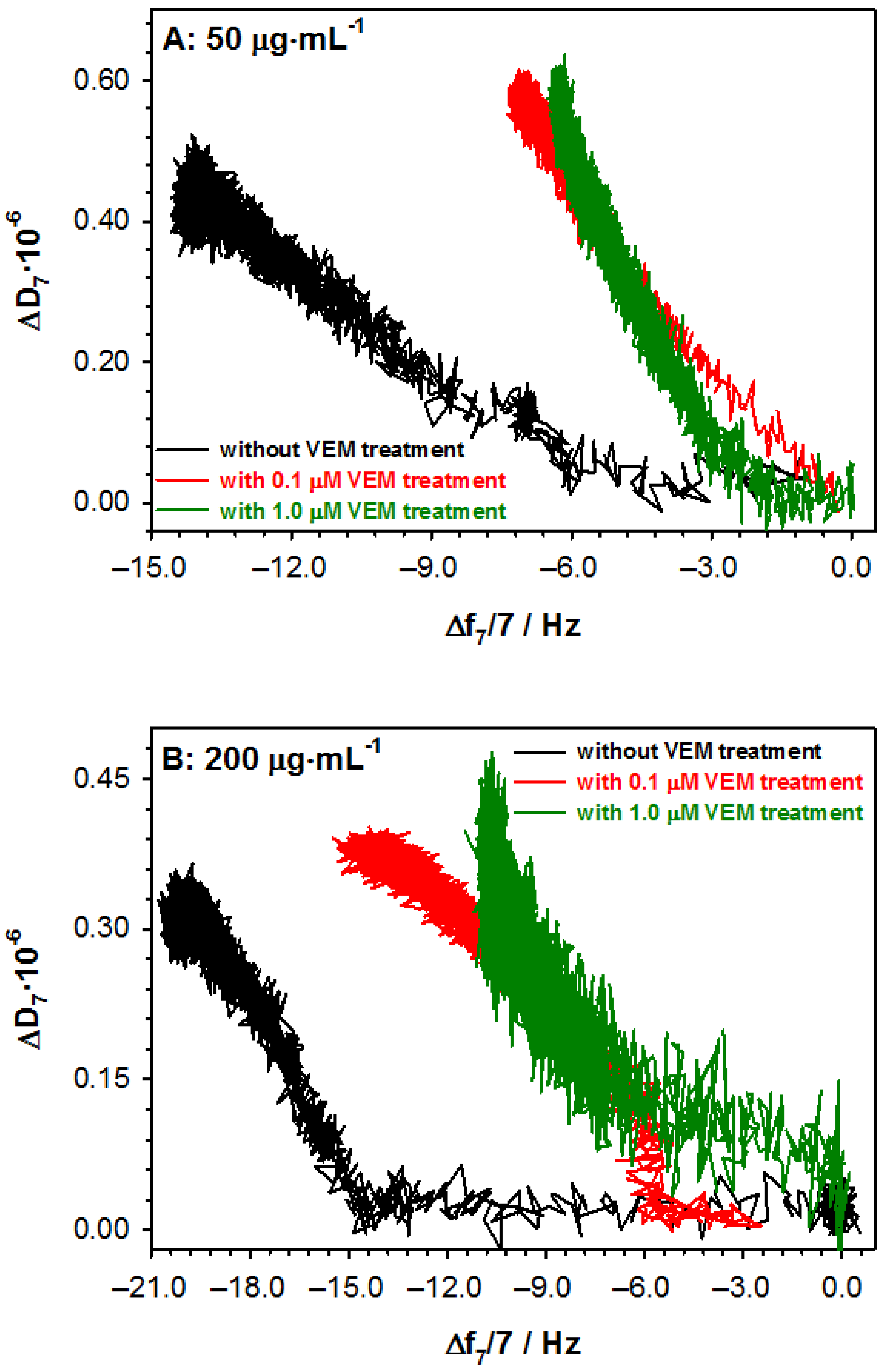
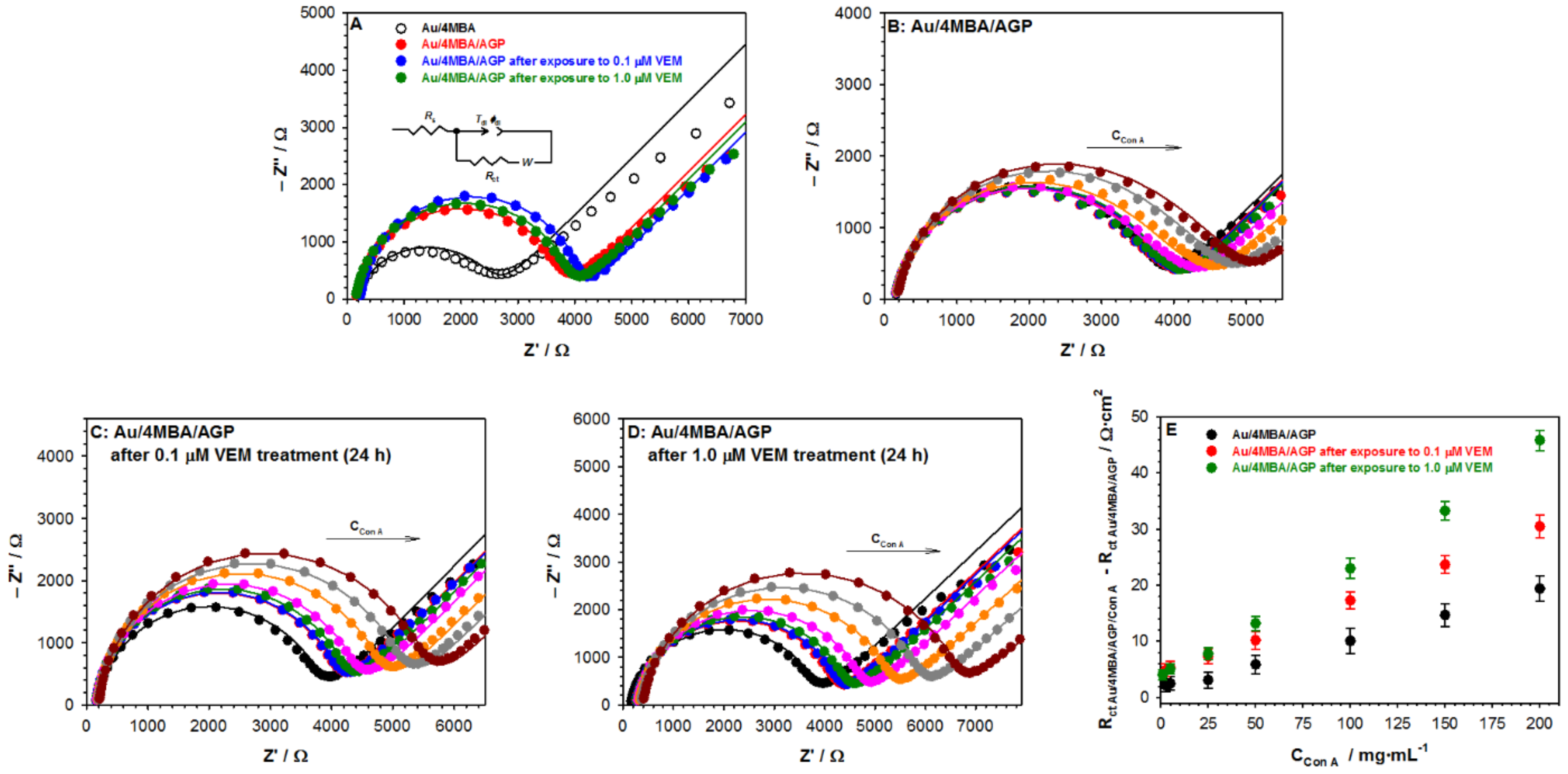
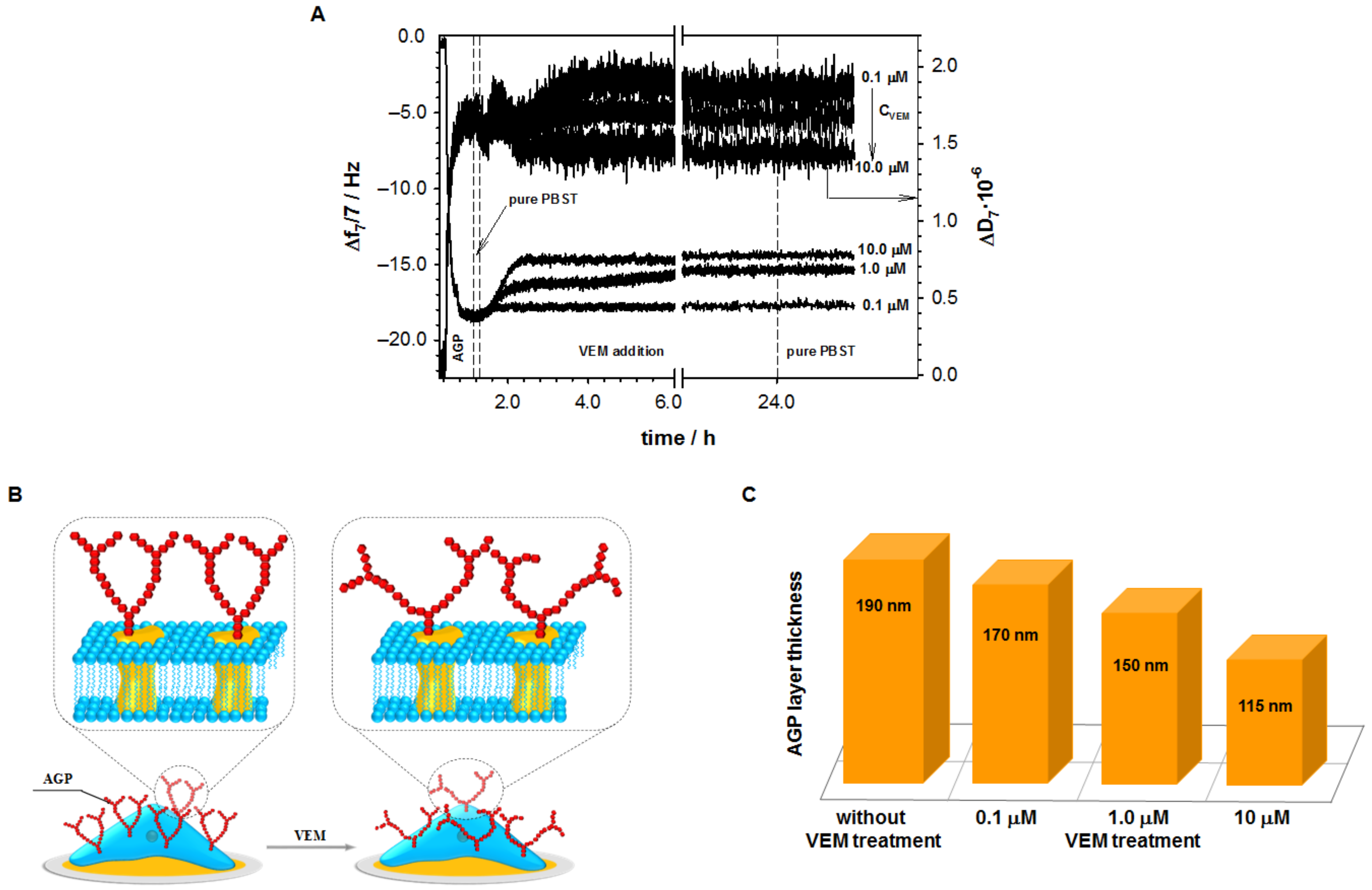
2.1. Influence of Vemurafenib on Glycoprotein Structure
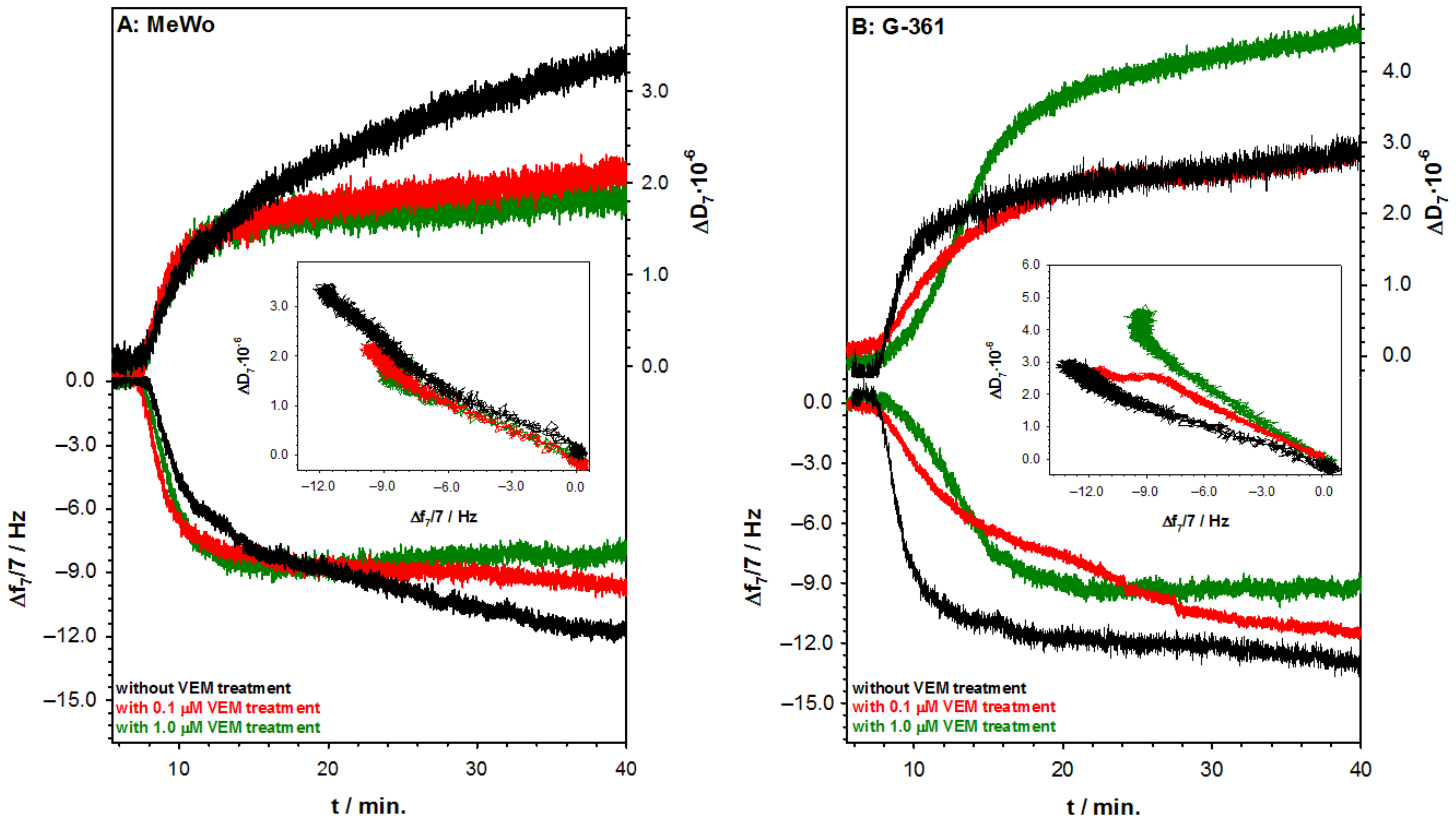
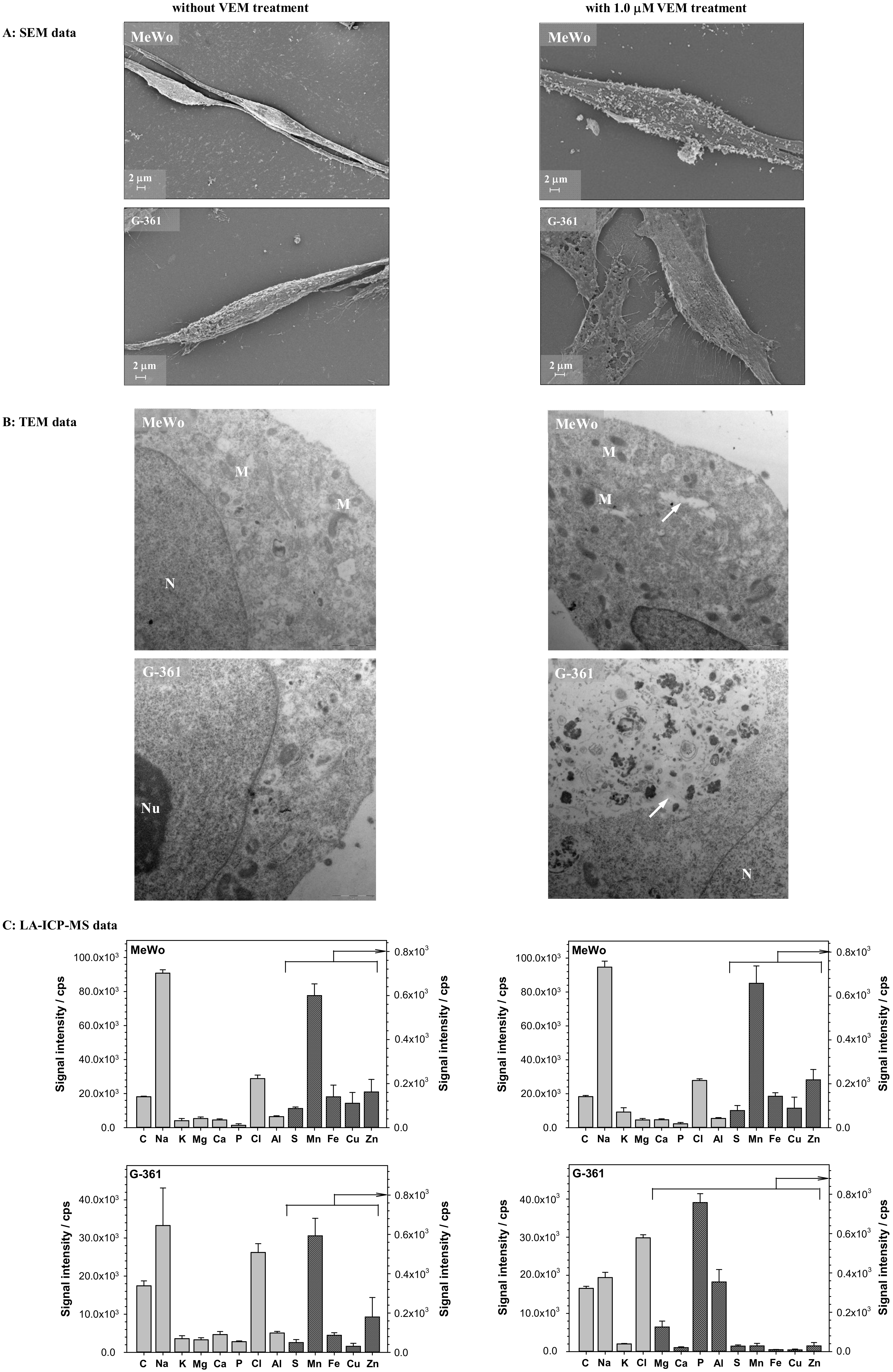
2.2. Studies with Melanoma Cell Lines without and with BRAF Mutation
3. Materials and Methods
3.1. Materials
3.2. Cell Culture
3.3. Quartz Crystal Microbalance with Dissipation (QCM-D)
3.4. Electrochemical Impedance Spectroscopy (EIS)
3.5. Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM) Imaging
3.6. ICP-MS Measurements with Laser Ablation (LA-ICP-MS)
3.7. Statistical Analysis
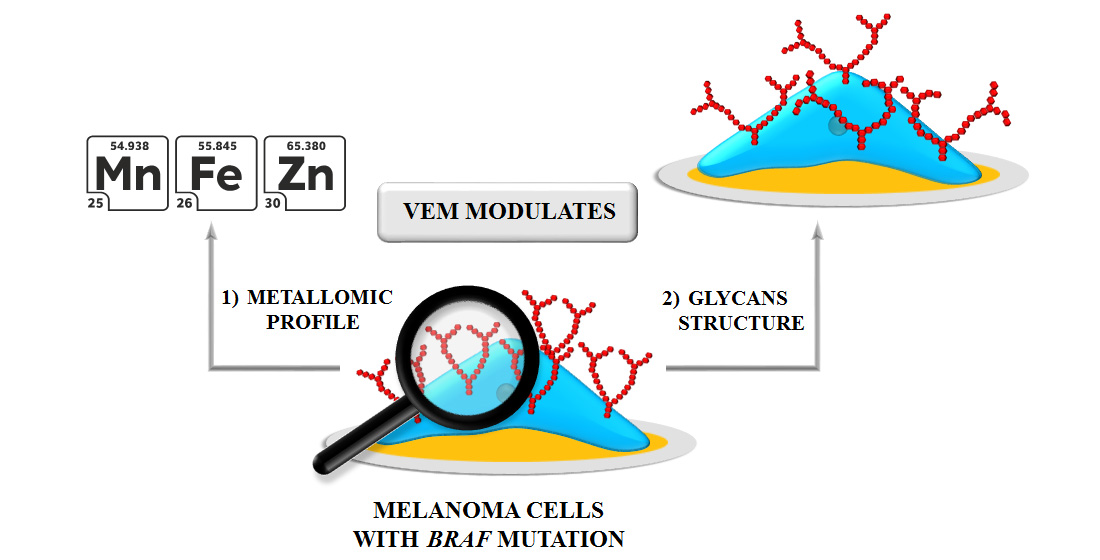
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGP | α1-acid glycoprotein |
| Con A | Canavalia ensiformis |
| DMSO | dimethyl sulfoxide |
| EIS | electrochemical impedance spectroscopy |
| LA-ICP-MS | inductively coupled plasma mass spectrometer with laser ablation |
| PBST | phosphate-buffered saline, 0.1% Tween 20 detergent |
| SEM | scanning electron microscopy |
| TEM | transmission electron microscopy |
| QCM-D | quartz crystal microbalance with dissipation |
| VEM | vemurafenib |
References
- Valverde, P.; Ardá, A.; Reichardt, N.-C.; Jimenez-Barbero, J.; Gimeno, A. Glycans in drug discovery. MedChemComm 2019, 10, 1678–1691. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.D.; Behan, J.; Matthews-Smith, G.; Magliocco, A.M. Alpha-1-Acid Glycoprotein (AGP) as a Potential Biomarker for Breast Cancer. In Glycosylation; Petrescu, S., Ed.; IntechOpen: London, UK, 2012; pp. 201–222. [Google Scholar]
- Schmid, K.; Nimberg, R.; Kimura, A.; Yamaguchi, H.; Binette, J. The carbohydrate units of human plasma α1-Acid glycoprotein. Biochim. Biophys. Acta Proteins Proteom. 1977, 492, 291–302. [Google Scholar] [CrossRef]
- Yoshima, H.; Matsumoto, A.; Mizuochi, T.; Kawasaki, T.; Kobata, A. Comparative study of the carbohydrate moieties of rat and human plasma alpha 1-acid glycoproteins. J. Biol. Chem. 1981, 256, 8476–8484. [Google Scholar] [PubMed]
- Bteich, M. An overview of albumin and alpha-1-acid glycoprotein main characteristics: Highlighting the roles of amino acids in binding kinetics and molecular interactions. Heliyon 2019, 5, e02879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budai, L.; Ozohanics, O.; Ludányi, K.; Drahos, L.; Kremmer, T.; Krenyácz, J.; Vékey, K. Investigation of genetic variants of α-1 acid glycoprotein by ultra-performance liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2008, 393, 991–998. [Google Scholar] [CrossRef]
- Kremmer, T.; Szöllosi, É.; Boldizsár, M.; Vincze, B.; Ludanyi, K.; Imre, T.; Schlosser, G.; Vekey, K. Liquid chromatographic and mass spectrometric analysis of human serum acid alpha-1-glycoprotein. Biomed. Chromatogr. 2004, 18, 323–329. [Google Scholar] [CrossRef]
- Ceciliani, F.; Pocacqua, V. The Acute Phase Protein α1-Acid Glycoprotein: A Model for Altered Glycosylation During Diseases. Curr. Protein Pept. Sci. 2007, 8, 91–108. [Google Scholar] [CrossRef]
- Imre, T.; Schlosser, G.; Pocsfalvi, G.; Siciliano, R.; Molnár-Szöllősi, É.; Kremmer, T.; Malorni, A.; Vékey, K.; Siciliano, R.A. Glycosylation site analysis of human alpha-1-acid glycoprotein (AGP) by capillary liquid chromatography—electrospray mass spectrometry. J. Mass Spectrom. 2005, 40, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Briard, J.G.; Jiang, H.; Moremen, K.W.; Macauley, M.S.; Wu, P. Cell-based glycan arrays for probing glycan–glycan binding protein interactions. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Dennis, J.W.; Nabi, I.R.; Demetriou, M. Metabolism, Cell Surface Organization, and Disease. Cell 2009, 139, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Senkara-Barwijuk, E.; Kobiela, T.; Lebed, K.; Lekka, M. Reaction pathway and free energy profile determined for specific recognition of oligosaccharide moiety of carboxypeptidase Y. Biosens. Bioelectron. 2012, 36, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Mazalovska, M.; Kouokam, J.C. Plant-Derived Lectins as Potential Cancer Therapeutics and Diagnostic Tools. BioMed Res. Int. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Naismith, J.H.; Emmerich, C.; Habash, J.; Harrop, S.J.; Helliwell, J.R.; Hunter, W.N.; Raftery, J.; Gilboa, A.J.K.; Yariv, J. Refined structure of concanavalin A complexed with methyl α-D-mannopyranoside at 2.0 Å resolution and comparison with the saccharide-free structure. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Poiroux, G.; Barre, A.; Van Damme, E.J.M.; Benoist, H.; Rougé, P. Plant Lectins Targeting O-Glycans at the Cell Surface as Tools for Cancer Diagnosis, Prognosis and Therapy. Int. J. Mol. Sci. 2017, 18, 1232. [Google Scholar] [CrossRef] [Green Version]
- Barre, A.; Bourne, Y.; Van Damme, E.J.M.; Rougé, P. Overview of the structure-function relationships of mannose-specific lectins from plants. Algae and Fungi. Int. J. Mol. Sci. 2019, 20, 254. [Google Scholar] [CrossRef] [Green Version]
- Sobiepanek, A.; Milner-Krawczyk, M.; Lekka, M.; Kobiela, T. AFM and QCM-D as tools for the distinction of melanoma cells with a different metastatic potential. Biosens. Bioelectron. 2017, 93, 274–281. [Google Scholar]
- Wang, W.; Antonsen, K.; Wang, Y.J.; Wang, D. pH dependent effect of glycosylation on protein stability. Eur. J. Pharm. Sci. 2008, 33, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Pažitná, L.; Nemčovič, M.; Pakanová, Z.; Baráth, P.; Aliev, T.; Dolgikh, D.; Argentova, V.; Katrlík, J. Influence of media composition on recombinant monoclonal IgA1 glycosylation analysed by lectin-based protein microarray and MALDI-MS. J. Biotechnol. 2020, 4, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Sajan, E.; Matanguihan, R.; Heidemann, R.; Abu-Absi, S.; Asuncion, W.; Yamasaki, G.; Wu, X.; Chen, J.; Murphy, J.E.; Zhang, C. The Effect of Bioreactor pH and Temperature on Protein Glycosylation in Perfusion Cultures of Mammalian Cells. Biotechnol. Bioeng. 2010, 94, 785–788. [Google Scholar] [CrossRef]
- Restelli, V.; Wang, M.-D.; Huzel, N.; Ethier, M.; Perreault, H.; Butler, M. The effect of dissolved oxygen on the production and the glycosylation profile of recombinant human erythropoietin produced from CHO cells. Biotechnol. Bioeng. 2006, 94, 481–494. [Google Scholar] [CrossRef]
- Wu, D.; Struwe, W.B.; Harvey, D.J.; Ferguson, M.A.J.; Robinson, C.V. N-glycan microheterogeneity regulates interactions of plasma proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 8763–8768. [Google Scholar] [CrossRef] [Green Version]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [Green Version]
- Reeke, G.N.; Becker, J.W.; Cunningham, B.A.; Wang, J.L.; Yahara, I.; Edelman, G.M. Structure and Function of Concanavalin A. Adv. Exp. Med. Biol. 1975, 55, 13–33. [Google Scholar] [CrossRef]
- Adamczyk, Z.; Weroński, P. Random sequential adsorption of spheroidal particles: Kinetics and jamming limit. J. Chem. Phys. 1996, 105, 5562–5573. [Google Scholar] [CrossRef] [Green Version]
- Kustermann, S.; Boess, F.; Buness, A.; Schmitz, M.; Watzele, M.; Weiser, T.; Singer, T.; Suter, L.; Roth, A. A label-free, impedance-based real time assay to identify drug-induced toxicities and differentiate cytostatic from cytotoxic effects. Toxicol. Vitr. 2013, 27, 1589–1595. [Google Scholar] [CrossRef]
- Jahnke, H.-G.; Heimann, A.; Azendorf, R.; Mpoukouvalas, K.; Kempski, O.; Robitzki, A.; Charalampaki, P. Impedance spectroscopy—An outstanding method for label-free and real-time discrimination between brain and tumor tissue in vivo. Biosens. Bioelectron. 2013, 46, 8–14. [Google Scholar] [CrossRef]
- Lei, K.-F.; Wu, M.-H.; Hsu, C.-W.; Chen, Y.-D. Real-time and non-invasive impedimetric monitoring of cell proliferation and chemosensitivity in a perfusion 3D cell culture microfluidic chip. Biosens. Bioelectron. 2014, 51, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, H.-G.; Poenick, S.; Maschke, J.; Kendler, M.; Simon, J.C.; Robitzki, A.A. Direct Chemosensitivity Monitoring Ex Vivo on Undissociated Melanoma Tumor Tissue by Impedance Spectroscopy. Cancer Res. 2014, 74, 6408–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. In vitro assays and techniques utilized in anticancer drug discovery. J. Appl. Toxicol. 2019, 39, 38–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpal, M.; Feala, J.; Puyang, X.; Zou, J.; Ramos, A.H.; Wu, J.; Baumeister, T.; Yu, L.; Warmuth, M.; Zhu, P. Implementation of In Vitro Drug Resistance Assays: Maximizing the Potential for Uncovering Clinically Relevant Resistance Mechanisms. J. Vis. Exp. 2015, 123, e52879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, D.; Rothe, R.; Kirsten, M.; Jahnke, H.-G.; Dumann, K.; Ziemer, M.; Simon, J.-C.; Robitzki, A. A multidimensional impedance platform for the real-time analysis of single and combination drug pharmacology in patient-derived viable melanoma models. Biosens. Bioelectron. 2019, 123, 185–194. [Google Scholar] [CrossRef]
- Nowacki, L.; Follet, J.; Vayssade, M.; Vigneron, P.; Rotellini, L.; Cambay, F.; Egles, C.; Rossi, C. Real-time QCM-D monitoring of cancer cell death early events in a dynamic context. Biosens. Bioelectron. 2015, 64, 469–476. [Google Scholar] [CrossRef]
- Tonda-Turo, C.; Carmagnola, I.; Ciardelli, G. Quartz Crystal Microbalance with Dissipation Monitoring: A Powerful Method to Predict the in vivo Behavior of Bioengineered Surfaces. Front. Bioeng. Biotechnol. 2018, 6, 158. [Google Scholar] [CrossRef]
- Migoń, D.; Wasilewski, T.; Suchy, D. Application of QCM in Peptide and Protein-Based Drug Product Development. Molecules 2020, 25, 3950. [Google Scholar] [CrossRef]
- He, Z.; Mansfeld, F. Exploring the use of electrochemical impedance spectroscopy (EIS) in microbial fuel cell studies. Energy Environ. Sci. 2008, 2, 215–219. [Google Scholar] [CrossRef]
- Hassan, Q.; Ahmadi, S.; Kerman, K. Recent Advances in Monitoring Cell Behavior Using Cell-Based Impedance Spectroscopy. Micromachines 2020, 11, 590. [Google Scholar] [CrossRef]
- Staniszewska, M.; Sobiepanek, A.; Gizińska, M.; Peña-Cabrera, E.; Arroyo-Córdoba, I.J.; Kazek, M.; Kuryk, Ł.; Wieczorek, M.; Koronkiewicz, M.; Kobiela, T.; et al. Sulfone derivatives enter the cytoplasm of Candida albicans sessile cells. Eur. J. Med. Chem. 2020, 191, 112139. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Tan, Z.; Chen, S.; Guan, F. Role of Glycans in Cancer Cells Undergoing Epithelial–Mesenchymal Transition. Front. Oncol. 2016, 6, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nat. Cell Biol. 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; McCusker, J.P.; Kluger, Y.; Sznol, M. PLX4032, a selective BRAFV600E kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAFWT melanoma cells. Pigment Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Zhu, L.; Lue, H.; Bernhagen, J.; Schwaneberg, U. Directed arginine deiminase evolution for efficient inhibition of arginine-auxotrophic melanomas. Appl. Microbiol. Biotechnol. 2014, 99, 1237–1247. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Chattopadhyay, C.; Akar, U.; Gabisi, A.; Newman, R.A.; Grimm, E.A. Zyflamend Mediates Therapeutic Induction of Autophagy to Apoptosis in Melanoma Cells. Nutr. Cancer 2011, 63, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 2017, 102, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; Pereira, G.J.S.; Saito, R.D.F.; Gil, C.D.; Zanatta, D.B.; Smaili, S.S.; Chammas, R. Galectin-3 sensitized melanoma cell lines to vemurafenib (PLX4032) induced cell death through prevention of autophagy. Oncotarget 2018, 9, 14567–14579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leo, L.; Bodemeyer, V.; De Zio, D. The Complex Role of Autophagy in Melanoma Evolution: New Perspectives from Mouse Models. Front. Oncol. 2020, 9, 1506. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-P.; Ambudkar, S.V. The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm. Sin. B 2014, 4, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Hoffmann, E.K. Role of Ion Transport in Control of Apoptotic Cell Death. Compr. Physiol. 2012, 2, 2037–2061. [Google Scholar] [CrossRef]
- Jiang, H.; López-Aguilar, A.; Meng, L.; Gao, Z.; Liu, Y.; Tian, X.; Yu, G.; Ovryn, B.; Moremen, K.W.; Wu, P. Modulating Cell-Surface Receptor Signaling and Ion Channel Functions by In Situ Glycan Editing. Angew. Chem. Int. Ed. 2018, 57, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Joseph, R.W.; Swaika, A.; Crozier, J.A. Vemurafenib: An evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2014, 8, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, J.G.; Liang, J.; Antonopoulos, A.; Giovannone, N.; Kang, S.; Mondala, T.S.; Head, S.R.; King, S.L.; Tani, Y.; Brackett, D.; et al. Loss of GCNT2/I-branched glycans enhances melanoma growth and survival. Nat. Commun. 2018, 9, 3368. [Google Scholar] [CrossRef] [PubMed]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Mallet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Goñi, T.; Miniotis, M.F.; Wantuch, S.; Parkes, H.G.; Marais, R.; Workman, P.; Leach, M.O.; Beloueche-Babari, M. The BRAF Inhibitor Vemurafenib Activates Mitochondrial Metabolism and Inhibits Hyperpolarized Pyruvate-Lactate Exchange in BRAF-Mutant Human Melanoma Cells. Mol. Cancer Ther. 2016, 15, 2987–2999. [Google Scholar] [CrossRef] [Green Version]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Gao, L.X.; Ma, X.Q.; Hu, F.X.; Li, C.M.; Lu, Z. Involvement of superoxide and nitric oxide in BRAFV600E inhibitor PLX4032-induced growth inhibition of melanoma cells. Integr. Biol. 2014, 6, 1211–1217. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting oncogenic BRAF: Past, present, and future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, E.S. The use of lead citrate at high pH as electron-opaque stain for electron microscopy. J. Cell Biol. 2004, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Modyfication | Tdl [μF·s(1−ϕ)·cm−2] | ϕdl | Rct [Ω·cm2] | σ [Ω·rad1/2·s−1/2·cm2] | Cdl [μF·cm−2] |
|---|---|---|---|---|---|
| Au | 82.1 ± 0.4 | 0.91 ± 0.02 | 6.5 ± 0.2 | 25.2 ± 0.3 | 23.0 ± 0.9 |
| Au/4MBA | 98.2 ± 2.2 | 0.87 ± 0.04 | 48.6 ± 2.2 | 39.0 ± 1.1 | 13.5 ± 0.4 |
| Au/4MBA/AGP | 33.4 ± 1.2 | 0.91 ± 0.03 | 72.8 ± 4.3 | 39.1 ± 2.5 | 12.8 ± 0.5 |
| Au/4MBA/AGPafter exposure to 0.1 μM VEM | 20.3 ± 3.5 | 0.92 ± 0.04 | 75.6 ± 5.2 | 38.5 ± 2.3 | 11.9 ± 1.0 |
| Au/4MBA/AGPafter exposure to 1.0 μM VEM | 20.4 ± 2.5 | 0.94 ± 0.03 | 77.8 ± 3.2 | 36.7 ± 2.4 | 9.0 ± 1.1 |
| Au/4MBA/AGP | Regression Equation | R2 | KA/[M−1] | KD/[M−1] |
|---|---|---|---|---|
| without VEM treatment | 0.998 | 3.82 × 106 | 2.61 × 10−7 | |
| with 0.1 μM VEM treatment | 0.991 | 2.42 × 106 | 4.13 × 10−7 | |
| with 1.0 μM VEM treatment | 0.997 | 1.34 × 106 | 7.46 × 10−7 |
| Concentartion of Con A [μg·mL−1] | Tdl [μF·s(1−ϕ)·cm−2] | ϕdl | Rct [Ω·cm2] | σ [Ω·rad1/2·s−1/2·cm2] | Cdl [μF·cm−2] |
|---|---|---|---|---|---|
| Au/4MBA/AGP | |||||
| 2.5 | 32.2 ± 3.6 | 0.88 ± 0.01 | 74.6 ± 2.7 | 28.8 ± 2.7 | 9.2 ± 0.8 |
| 5.0 | 32.0 ± 2.3 | 0.88 ± 0.02 | 75.2 ± 1.2 | 29.0 ± 2.2 | 9.2 ± 0.6 |
| 25 | 31.8 ± 2.7 | 0.88 ± 0.01 | 75.6 ± 1.4 | 29.2 ± 2.1 | 9.1 ± 0.8 |
| 50 | 30.5 ± 1.3 | 0.88 ± 0.03 | 78.6 ± 2.6 | 30.3 ± 2.5 | 8.7 ± 0.4 |
| 100 | 28.8 ± 2.6 | 0.89 ± 0.02 | 83.4 ± 2.7 | 32.2 ± 2.6 | 9.2 ± 0.7 |
| 150 | 27.2 ± 1.6 | 0.89 ± 0.02 | 88.5 ± 2.7 | 34.1 ± 2.6 | 8.8 ± 0.7 |
| 200 | 25.9 ± 2.3 | 0.89 ± 0.02 | 92.9 ± 2.7 | 35.8 ± 2.2 | 8.4 ± 0.8 |
| Au/4MBA/AGP after exposure to 0.1 μM VEM | |||||
| 2.5 | 22.6 ± 1.6 | 0.94 ± 0.01 | 78.4 ± 2.7 | 46.9 ± 2.7 | 12.5 ± 0.8 |
| 5.0 | 22.5 ± 2.3 | 0.94 ± 0.02 | 78.8 ± 1.2 | 47.1 ± 3.2 | 12.5 ± 0.6 |
| 25 | 21.8 ± 1.7 | 0.94 ± 0.01 | 81.0 ± 1.4 | 48.4 ± 2.1 | 12.1 ± 0.8 |
| 50 | 21.0 ± 1.3 | 0.95 ± 0.03 | 84.2 ± 3.6 | 50.4 ± 2.5 | 12.8 ± 0.4 |
| 100 | 19.3 ± 1.6 | 0.95 ± 0.02 | 91.9 ± 2.7 | 54.9 ± 2.6 | 11.8 ± 0.7 |
| 150 | 18.0 ± 1.6 | 0.95 ± 0.02 | 98.7 ± 2.7 | 58.9 ± 2.6 | 11.0 ± 0.7 |
| 200 | 16.8 ± 1.3 | 0.95 ± 0.02 | 105.7 ± 5.7 | 63.2 ± 2.2 | 10.3 ± 0.8 |
| Au/4MBA/AGP after exposure to 1.0 μM VEM | |||||
| 2.5 | 25.0 ± 1.6 | 0.92 ± 0.01 | 79.6 ± 2.7 | 37.2 ± 2.7 | 11.5 ± 0.8 |
| 5.0 | 24.7 ± 1.3 | 0.92 ± 0.02 | 80.6 ± 3.2 | 37.6 ± 3.2 | 11.5 ± 0.6 |
| 25 | 23.8 ± 1.7 | 0.92 ± 0.01 | 83.4 ± 2.4 | 39.0 ± 3.1 | 11.0 ± 0.8 |
| 50 | 22.5 ± 1.3 | 0.93 ± 0.03 | 89.3 ± 2.6 | 41.8 ± 2.5 | 11.4 ± 0.4 |
| 100 | 20.0 ± 1.6 | 0.93 ± 0.02 | 100.1 ± 2.7 | 46.7 ± 2.6 | 10.3 ± 0.7 |
| 150 | 18.0 ± 1.6 | 0.94 ± 0.02 | 110.9 ± 2.7 | 51.9 ± 2.6 | 10.2 ± 0.7 |
| 200 | 16.0 ± 1.3 | 0.95 ± 0.02 | 124.6 ± 4.7 | 58.3 ± 1.2 | 9.5 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisiewicz, M.K.; Kowalczyk, A.; Sobiepanek, A.; Jagielska, A.; Wagner, B.; Nowakowska, J.; Gniadek, M.; Grudzinski, I.P.; Kobiela, T.; Nowicka, A.M. Tracking of Glycans Structure and Metallomics Profiles in BRAF Mutated Melanoma Cells Treated with Vemurafenib. Int. J. Mol. Sci. 2021, 22, 439. https://doi.org/10.3390/ijms22010439
Nisiewicz MK, Kowalczyk A, Sobiepanek A, Jagielska A, Wagner B, Nowakowska J, Gniadek M, Grudzinski IP, Kobiela T, Nowicka AM. Tracking of Glycans Structure and Metallomics Profiles in BRAF Mutated Melanoma Cells Treated with Vemurafenib. International Journal of Molecular Sciences. 2021; 22(1):439. https://doi.org/10.3390/ijms22010439
Chicago/Turabian StyleNisiewicz, Monika K., Agata Kowalczyk, Anna Sobiepanek, Agata Jagielska, Barbara Wagner, Julita Nowakowska, Marianna Gniadek, Ireneusz P. Grudzinski, Tomasz Kobiela, and Anna M. Nowicka. 2021. "Tracking of Glycans Structure and Metallomics Profiles in BRAF Mutated Melanoma Cells Treated with Vemurafenib" International Journal of Molecular Sciences 22, no. 1: 439. https://doi.org/10.3390/ijms22010439