Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy



Abstract
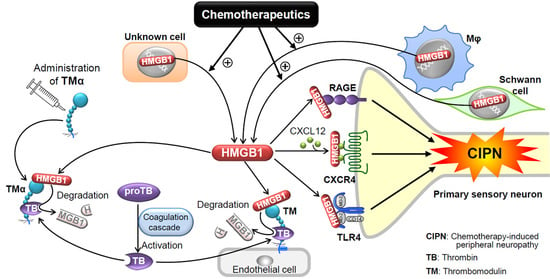
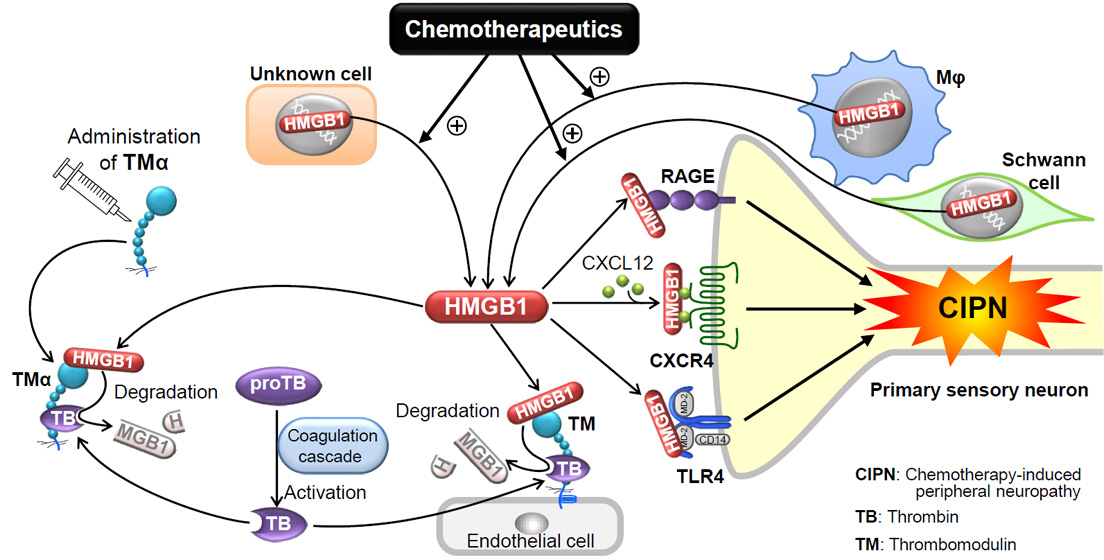
1. Introduction
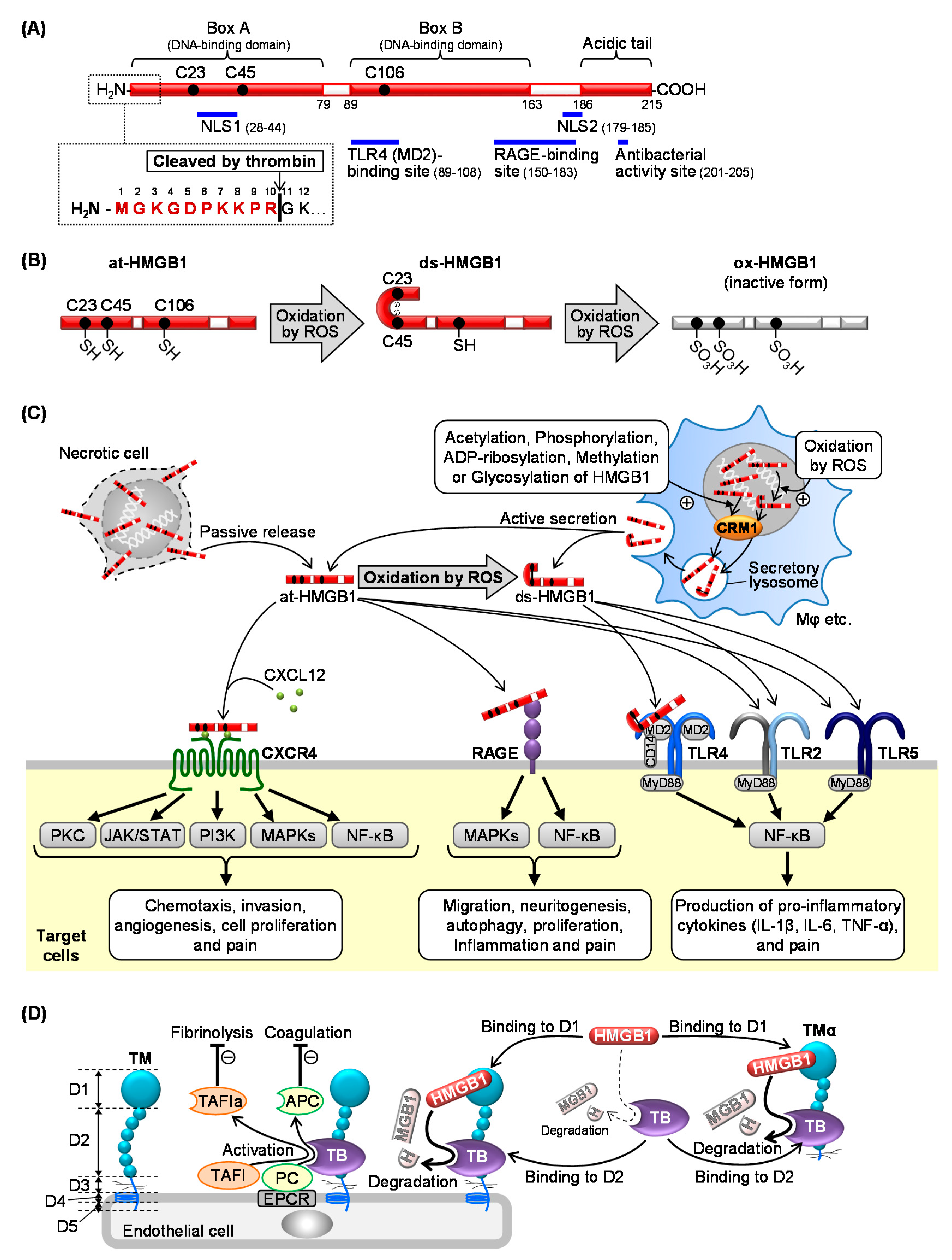
2. Molecular and Biological Characteristics of HMGB1
2.1. Structure of HMGB1
2.2. Release of HMGB1
2.3. Membrane Receptors of Extracellular HMGB1 Involved in Pain Processing
2.3.1. RAGE
2.3.2. TLRs
2.3.3. CXCR4
2.4. Inactivation of HMGB1 by the Thrombin and TM System
3. Role of HMGB1 in CIPN
3.1. Involvement of Endogenous HMGB1 in the Development of Pathological Pain Including CIPN
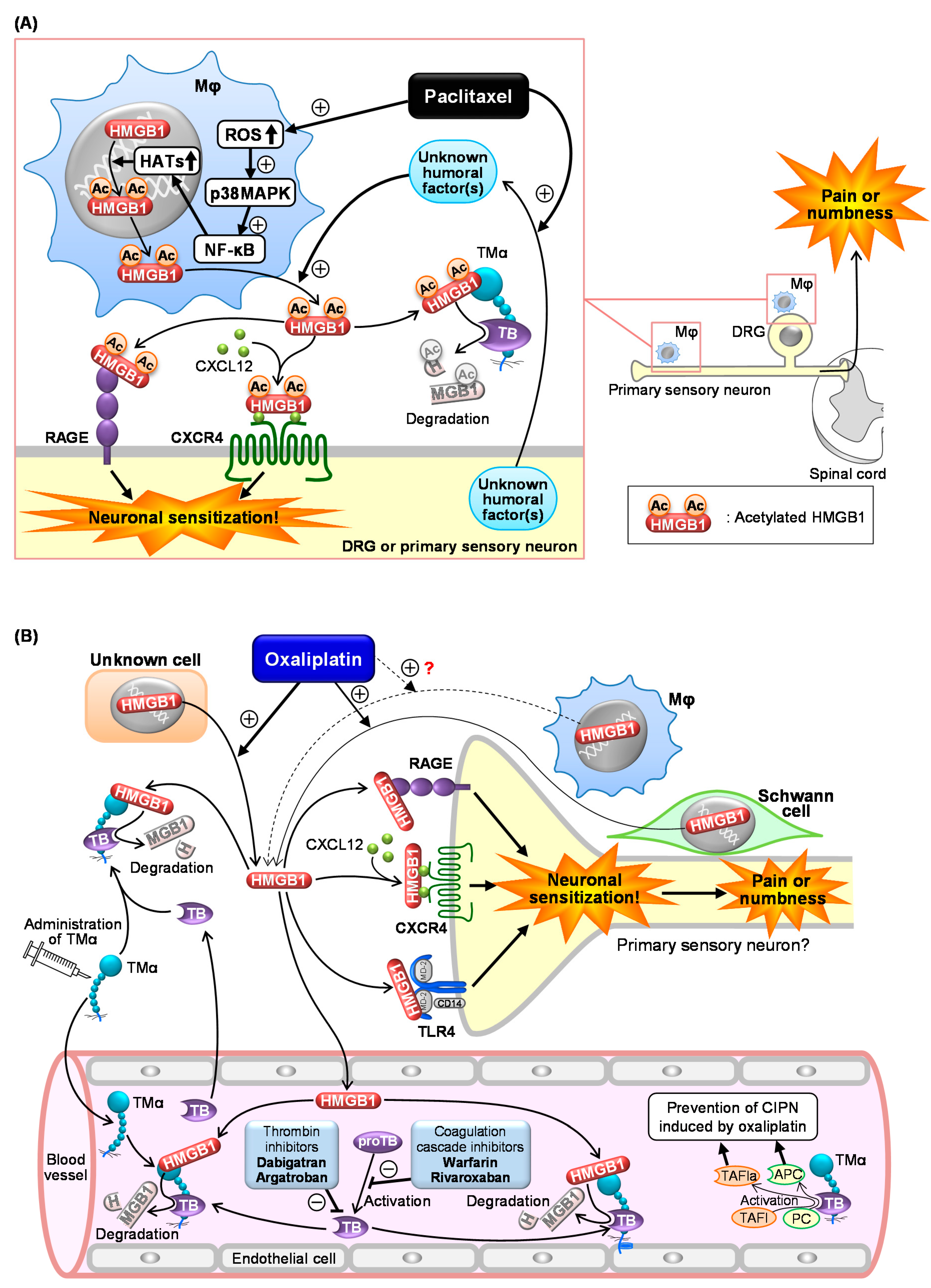
3.2. Roles of Macrophage-Derived HMGB1 and Its Upstream/Downstream Molecules in CIPN Caused by Paclitaxel
3.3. Role of HMGB1 and Its Upstream/Downstream Molecules in CIPN Caused by Oxaliplatin
4. Inactivation of HMGB1 for Prevention of CIPN
4.1. CIPN Prevention by Neutralization of HMGB1 with Monoclonal Antibodies
4.2. Thrombin-Dependent Suppression of CIPN by Endogenous and Exogenous TM
4.3. Other Candidates that Directly Inactivate HMGB1
5. Blocking Membrane Receptors of HMGB1 for Prevention of CIPN
5.1. RAGE Antagonists
5.2. TLR Antagonists
5.3. CXCR4 Antagonists
6. Targeting Macrophages for Prevention of CIPN Due to Paclitaxel

{kind=link}
{kind=link}
{kind=link}
| Chemicals (Alternative Name) | Structure | Mechanisms of Action | Ref. | Prevention of CIPN (Administration Route; Animal; Anticancer Drug) | Ref. |
|---|---|---|---|---|---|
| HMGB1-nAb | IgG (MW 150,000) | Binding to and inactivation of HMGB1 | [21,22] | Yes (i.p.; rat; PCT, VCR) Yes (i.p.; mouse; PCT) Yes (i.p.; mouse; OHP) | [8] [9] [10] |
| TMα | Protein (MW 64,000) | Binding to HMGB1 and promotion of degradation of HMGB1 by thrombin | [18,35,36,89] | Yes (i.p.; rat; PCT, VCR) Yes (i.p.; mouse; PCT) Yes (i.p., mouse; OHP) Yes (i.v.; rat; OHP) Yes (i.v.; human; OHP) | [8] [9] [10] [101] [37] |
| Glycyrrhizin (glycyrrhizic acid) | Small molecule (MW 822.9) | Binding to both box A and box B of HMGB1 | [104] | N.D. | |
| Methotrexate | Small molecule (MW 454.44) | Binding to HMGB1 and inhibition of HMGB1/RAGE interaction | [111] | N.D. | |
| Metformin | Small molecule (MW 129.16) | Binding to the C-terminal acidic tail of HMGB1 | [112] | Yes (i.p.; rat; OHP) Yes (i.p.; mouse; CDDP) | [116] [115] |
| EGCG | A green tea component, small molecule (MW 458.37) | Binding to HMGB1 and induction of aggregation of HMGB1 | [119] | N.D. | |
| Salicylic acid | Aspirin metabolite, small molecule (MW 138.12) | Bonding to box A and box B of HMGB1 | [120] | N.D. | |
| FPS-ZM1 | Small molecule (MW 327.8) | Blockade of RAGE | [67,121] | Yes (i.p.; mouse; PCT) Yes (i.p.; mouse; OHP) | [9] [10] |
| LMWH (parnaparin) | (MW 4500~6500) | Blockade of RAGE | [122,123] | Yes (i.p.; mouse; PCT) Yes (i.p.; mouse; OHP) | [9] [10] |
| Azeliragon (PF-04494700 or TTP488) | Small molecule (MW 532.1) | Blockade of RAGE | [67,124,125,126] | N.D. | |
| LPS-RS | LPS from the photosynthetic bacterium Rhodobacter sphaeroides | Blockade of TLR4 | [127,139] | Yes (i.t.; rat; PCT) No (i.p.; mouse; PCT) Yes (i.p.; mouse; OHP) | [95] [9] [10] |
| Eritoran (E5564) | Synthetic lipid A analogue (MW 1313.7) | Blockade of TLR4 | [127,131] | N.D. | |
| FP7 | Synthetic monosaccharide lipid A mimetic with about half MW of eritoran | Blockade of TLR4 | [132,133] | N.D. | |
| TAK-242 (CLI-095) | Small molecule (MW 361.82) | Blockade of TLR4 | [134,135] | Yes (i.v.; rat; PCT) Yes (i.p.; mouse *; PCT) No (i.p.; mouse **; PCT) Yes (i.p.; mouse, OHP) | [94] [9] [9] [10] |
| AMD3100 (plerixafor) | Small bicyclam molecule (MW 129.16) | Blockade of CXCR4 | [136] | Yes (i.p.; mouse; PCT) Yes (i.p.; mouse; OHP) | [9] [10] |
| Ethyl pyruvate | Stable lipophilic pyruvate derivative, small molecule (MW 116.11) | Inhibition of HMGB1 secretion from macrophage | [137] | Yes (i.p.; mouse; PCT) No (i.p.; mouse; OHP) | [9] [10] |
| Minocycline | Small molecule (MW 457.48) | Inhibition of macrophage/microglia | [138] | Yes (i.p.; mouse; PCT) No (i.p.; mouse; OHP) | [9] [10] |
| NAC | Small molecule (MW 163.20) | Anti-oxidation, suppression of HMGB1 release | [9,55] | Yes (i.p.; mouse; PCT) | [9] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3325–3348. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J.L.; Dougherty, P.M.; Colvin, L.A. Clinical and preclinical perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): A narrative review. Br. J. Anaesth. 2017, 119, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Makker, P.G.; Tonkin, R.S.; Abdulla, M.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 2017, 73, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Trecarichi, A.; Flatters, S.J.L. Mitochondrial dysfunction in the pathogenesis of chemotherapy-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 83–126. [Google Scholar] [PubMed]
- Shim, H.S.; Bae, C.; Wang, J.; Lee, K.H.; Hankerd, K.M.; Kim, H.K.; Chung, J.M.; La, J.H. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol. Pain 2019, 15, 1744806919840098. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kaneko, S. Roles of Transient Receptor Potential Ankyrin 1 in Oxaliplatin-Induced Peripheral Neuropathy. Biol. Pharm. Bull. 2017, 40, 947–953. [Google Scholar] [CrossRef]
- Tomita, S.; Sekiguchi, F.; Deguchi, T.; Miyazaki, T.; Ikeda, Y.; Tsubota, M.; Yoshida, S.; Nguyen, H.D.; Okada, T.; Toyooka, N.; et al. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology 2019, 413, 33–39. [Google Scholar] [CrossRef]
- Nishida, T.; Tsubota, M.; Kawaishi, Y.; Yamanishi, H.; Kamitani, N.; Sekiguchi, F.; Ishikura, H.; Liu, K.; Nishibori, M.; Kawabata, A. Involvement of high mobility group box 1 in the development and maintenance of chemotherapy-induced peripheral neuropathy in rats. Toxicology 2016, 365, 48–58. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Domoto, R.; Nakashima, K.; Yamasoba, D.; Yamanishi, H.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacology 2018, 141, 201–213. [Google Scholar] [CrossRef]
- Tsubota, M.; Fukuda, R.; Hayashi, Y.; Miyazaki, T.; Ueda, S.; Yamashita, R.; Koike, N.; Sekiguchi, F.; Wake, H.; Wakatsuki, S.; et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: Negative impact of anticoagulants. J. Neuroinflamm. 2019, 16, 199. [Google Scholar] [CrossRef]
- Tsujita, R.; Tsubota, M.; Sekiguchi, F.; Kawabata, A. Role of high-mobility group box 1 and its modulation by thrombomodulin/thrombin axis in neuropathic and inflammatory pain. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Aspects Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtlander, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Taniguchi, T. High-mobility group box family of proteins: Ligand and sensor for innate immunity. Trends Immunol. 2012, 33, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Khanabdali, R.; Kalionis, B.; Tai, X.; Xia, S. The Emerging Role of HMGB1 in Neuropathic Pain: A Potential Therapeutic Target for Neuroinflammation. J. Immunol. Res. 2016, 2016, 6430423. [Google Scholar] [CrossRef]
- Yamasoba, D.; Tsubota, M.; Domoto, R.; Sekiguchi, F.; Nishikawa, H.; Liu, K.; Nishibori, M.; Ishikura, H.; Yamamoto, T.; Taga, A.; et al. Peripheral HMGB1-induced hyperalgesia in mice: Redox state-dependent distinct roles of RAGE and TLR4. J. Pharmacol. Sci. 2016, 130, 139–142. [Google Scholar] [CrossRef]
- VanPatten, S.; Al-Abed, Y. High Mobility Group Box-1 (HMGb1): Current Wisdom and Advancement as a Potential Drug Target. J. Med. Chem. 2018, 61, 5093–5107. [Google Scholar] [CrossRef]
- Tsujita, R.; Tsubota, M.; Hayashi, Y.; Saeki, H.; Sekiguchi, F.; Kawabata, A. Role of Thrombin in Soluble Thrombomodulin-Induced Suppression of Peripheral HMGB1-Mediated Allodynia in Mice. J. Neuroimmune. Pharmacol. 2018, 13, 179–188. [Google Scholar] [CrossRef]
- Hiramoto, S.; Tsubota, M.; Yamaguchi, K.; Okazaki, K.; Sakaegi, A.; Toriyama, Y.; Tanaka, J.; Sekiguchi, F.; Ishikura, H.; Wake, H.; et al. Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice. Cells 2020, 9, 1748. [Google Scholar] [CrossRef]
- Irie, Y.; Tsubota, M.; Maeda, M.; Hiramoto, S.; Sekiguchi, F.; Ishikura, H.; Wake, H.; Nishibori, M.; Kawabata, A. HMGB1 and its membrane receptors as therapeutic targets in an intravesical substance P-induced bladder pain syndrome mouse model. J. Pharmacol. Sci. 2020, 143, 112–116. [Google Scholar] [CrossRef]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef]
- Shibasaki, M.; Sasaki, M.; Miura, M.; Mizukoshi, K.; Ueno, H.; Hashimoto, S.; Tanaka, Y.; Amaya, F. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain 2010, 149, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Wang, W.; Huang, J.; Ren, N.; Wu, S.X.; Li, Y.Q. Spinal high-mobility group box 1 contributes to mechanical allodynia in a rat model of bone cancer pain. Biochem. Biophys. Res. Commun. 2010, 395, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Otoshi, K.; Kikuchi, S.; Kato, K.; Sekiguchi, M.; Konno, S. Anti-HMGB1 neutralization antibody improves pain-related behavior induced by application of autologous nucleus pulposus onto nerve roots in rats. Spine 2011, 36, E692–E698. [Google Scholar] [CrossRef]
- Ren, P.C.; Zhang, Y.; Zhang, X.D.; An, L.J.; Lv, H.G.; He, J.; Gao, C.J.; Sun, X.D. High-mobility group box 1 contributes to mechanical allodynia and spinal astrocytic activation in a mouse model of type 2 diabetes. Brain Res. Bull. 2012, 88, 332–337. [Google Scholar] [CrossRef]
- Nakamura, Y.; Morioka, N.; Abe, H.; Zhang, F.F.; Hisaoka-Nakashima, K.; Liu, K.; Nishibori, M.; Nakata, Y. Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS ONE 2013, 8, e73640. [Google Scholar] [CrossRef]
- Tanaka, J.; Seki, Y.; Ishikura, H.; Tsubota, M.; Sekiguchi, F.; Yamaguchi, K.; Murai, A.; Umemura, T.; Kawabata, A. Recombinant human soluble thrombomodulin prevents peripheral HMGB1-dependent hyperalgesia in rats. Br. J. Pharmacol. 2013, 170, 1233–1241. [Google Scholar] [CrossRef]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundback, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef]
- Tanaka, J.; Yamaguchi, K.; Ishikura, H.; Tsubota, M.; Sekiguchi, F.; Seki, Y.; Tsujiuchi, T.; Murai, A.; Umemura, T.; Kawabata, A. Bladder pain relief by HMGB1 neutralization and soluble thrombomodulin in mice with cyclophosphamide-induced cystitis. Neuropharmacology 2014, 79, 112–118. [Google Scholar] [CrossRef]
- Zhang, F.F.; Morioka, N.; Harano, S.; Nakamura, Y.; Liu, K.; Nishibori, M.; Hisaoka-Nakashima, K.; Nakata, Y. Perineural expression of high-mobility group box-1 contributes to long-lasting mechanical hypersensitivity via matrix metalloprotease-9 up-regulation in mice with painful peripheral neuropathy. J. Neurochem. 2016, 136, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Tsubota, M.; Ishikura, H.; Sekiguchi, F.; Terada, Y.; Tsujiuchi, T.; Liu, K.; Nishibori, M.; Kawabata, A. Macrophage-derived HMGB1 as a Pain Mediator in the Early Stage of Acute Pancreatitis in Mice: Targeting RAGE and CXCL12/CXCR4 Axis. J. Neuroimmune. Pharmacol. 2017, 12, 693–707. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Rong, H.; Ni, H.; Zhu, C.; Xu, L.; Liu, Q.; Chen, Y.; Zheng, Y.; Huang, B.; Yao, M. Spinal PKC activation-Induced neuronal HMGB1 translocation contributes to hyperalgesia in a bone cancer pain model in rats. Exp. Neurol. 2018, 303, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, W.; Harada, S.; Liu, K.; Nishibori, M.; Tokuyama, S. Evidence of a role for spinal HMGB1 in ischemic stress-induced mechanical allodynia in mice. Brain Res. 2018, 1687, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abeyama, K.; Stern, D.M.; Ito, Y.; Kawahara, K.; Yoshimoto, Y.; Tanaka, M.; Uchimura, T.; Ida, N.; Yamazaki, Y.; Yamada, S.; et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J. Clin. Investig. 2005, 115, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kawahara, K.; Okamoto, K.; Yamada, S.; Yasuda, M.; Imaizumi, H.; Nawa, Y.; Meng, X.; Shrestha, B.; Hashiguchi, T.; et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1825–1830. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Saito, Y.; Kato, T.; Satake, H.; Makiyama, A.; Tsuji, Y.; Shinozaki, K.; Fujiwara, T.; Mizushima, T.; Harihara, Y.; et al. A placebo-controlled, double-blind, randomized study of recombinant thrombomodulin (ART-123) to prevent oxaliplatin-induced peripheral neuropathy. Cancer Chemother. Pharmacol. 2020, 86, 607–618. [Google Scholar] [CrossRef]
- Gong, W.; Li, Y.; Chao, F.; Huang, G.; He, F. Amino acid residues 201-205 in C-terminal acidic tail region plays a crucial role in antibacterial activity of HMGB1. J. Biomed. Sci. 2009, 16, 83. [Google Scholar] [CrossRef]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar]
- Li, J.; Kokkola, R.; Tabibzadeh, S.; Yang, R.; Ochani, M.; Qiang, X.; Harris, H.E.; Czura, C.J.; Wang, H.; Ulloa, L.; et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol. Med. 2003, 9, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Chuan-Xin, W.; Hang, S.; Qi, L.; Hui, G.; Jian-Ping, G. LPS induces HMGB1 relocation and release by activating the NF-κB-CBP signal transduction pathway in the murine macrophage-like cell line RAW264.7. J. Surg. Res. 2012, 175, 88–100. [Google Scholar]
- Xu, B.; Lang, L.M.; Lian, S.; Guo, J.R.; Wang, J.F.; Liu, J.; Yang, H.M.; Li, S.Z. Neuroinflammation induced by secretion of acetylated HMGB1 from activated microglia in hippocampi of mice following chronic cold exposure. Brain Res. 2020, 1726, 146495. [Google Scholar] [CrossRef]
- Peng, H.H.; Liu, Y.J.; Ojcius, D.M.; Lee, C.M.; Chen, R.H.; Huang, P.R.; Martel, J.; Young, J.D. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci. Rep. 2017, 7, 16628. [Google Scholar] [CrossRef]
- Parodi, M.; Pedrazzi, M.; Cantoni, C.; Averna, M.; Patrone, M.; Cavaletto, M.; Spertino, S.; Pende, D.; Balsamo, M.; Pietra, G.; et al. Natural Killer (NK)/melanoma cell interaction induces NK-mediated release of chemotactic High Mobility Group Box-1 (HMGB1) capable of amplifying NK cell recruitment. Oncoimmunology 2015, 4, e1052353. [Google Scholar] [CrossRef]
- Alsousi, A.A.; Igwe, O.J. Redox-active trace metal-induced release of high mobility group box 1(HMGB1) and inflammatory cytokines in fibroblast-like synovial cells is Toll-like receptor 4 (TLR4) dependent. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3847–3858. [Google Scholar] [CrossRef]
- Chi, J.H.; Seo, G.S.; Cheon, J.H.; Lee, S.H. Isoliquiritigenin inhibits TNF-alpha-induced release of high-mobility group box 1 through activation of HDAC in human intestinal epithelial HT-29 cells. Eur. J. Pharmacol. 2017, 796, 101–109. [Google Scholar] [CrossRef]
- Kang, L.; Guo, N.; Liu, X.; Wang, X.; Guo, W.; Xie, S.M.; Liu, C.; Lv, P.; Xing, L.; Zhang, X.; et al. High mobility group box-1 protects against Aflatoxin G1-induced pulmonary epithelial cell damage in the lung inflammatory environment. Toxicol. Lett. 2020, 331, 92–101. [Google Scholar] [CrossRef]
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 2018, 8, 2068. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J.; et al. LPS Induces Active HMGB1 Release From Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.; Song, J.; Lan, T.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Cardiomyocyte-fibroblast interaction contributes to diabetic cardiomyopathy in mice: Role of HMGB1/TLR4/IL-33 axis. Biochim. Biophys. Acta 2015, 1852, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pisetsky, D.S. The role of IFN-alpha and nitric oxide in the release of HMGB1 by RAW 264.7 cells stimulated with polyinosinic-polycytidylic acid or lipopolysaccharide. J. Immunol. 2006, 177, 3337–3343. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef]
- Loukili, N.; Rosenblatt-Velin, N.; Li, J.; Clerc, S.; Pacher, P.; Feihl, F.; Waeber, B.; Liaudet, L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc. Res. 2011, 89, 586–594. [Google Scholar] [CrossRef]
- Rendon-Mitchell, B.; Ochani, M.; Li, J.; Han, J.; Wang, H.; Yang, H.; Susarla, S.; Czura, C.; Mitchell, R.A.; Chen, G.; et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J. Immunol. 2003, 170, 3890–3897. [Google Scholar] [CrossRef]
- Toki, Y.; Takenouchi, T.; Harada, H.; Tanuma, S.; Kitani, H.; Kojima, S.; Tsukimoto, M. Extracellular ATP induces P2X7 receptor activation in mouse Kupffer cells, leading to release of IL-1beta, HMGB1, and PGE2, decreased MHC class I expression and necrotic cell death. Biochem. Biophys. Res. Commun. 2015, 458, 771–776. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, Y.M.; Kim, H.J.; Chang, K.C. Degradation of histone deacetylase 4 via the TLR4/JAK/STAT1 signaling pathway promotes the acetylation of high mobility group box 1 (HMGB1) in lipopolysaccharide-activated macrophages. FEBS Open Bio 2018, 8, 1119–1126. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, I.D.; Kim, S.W.; Lee, H.K.; Jin, Y.; Park, J.H.; Kim, T.K.; Suh, C.K.; Kwak, J.; Lee, K.H.; et al. Ethyl pyruvate inhibits HMGB1 phosphorylation and release by chelating calcium. Mol. Med. 2015, 20, 649–657. [Google Scholar] [CrossRef]
- Li, Y.; Xie, J.; Li, X.; Fang, J. Poly (ADP-ribosylation) of HMGB1 facilitates its acetylation and promotes HMGB1 translocation-associated chemotherapy-induced autophagy in leukaemia cells. Oncol. Lett. 2020, 19, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Fukazawa, J.; Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 2007, 282, 16336–16344. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kwak, M.S.; Park, J.B.; Lee, S.A.; Choi, J.E.; Cho, H.S.; Shin, J.S. N-linked glycosylation plays a crucial role in the secretion of HMGB1. J. Cell Sci. 2016, 129, 29–38. [Google Scholar] [CrossRef]
- Kwak, M.S.; Kim, H.S.; Lee, B.; Kim, Y.H.; Son, M.; Shin, J.S. Immunological Significance of HMGB1 Post-Translational Modification and Redox Biology. Front. Immunol. 2020, 11, 1189. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer-a Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- El-Far, A.H.; Sroga, G.; Jaouni, S.K.A.; Mousa, S.A. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 3613. [Google Scholar] [CrossRef]
- Zhong, H.; Li, X.; Zhou, S.; Jiang, P.; Liu, X.; Ouyang, M.; Nie, Y.; Chen, X.; Zhang, L.; Liu, Y.; et al. Interplay between RAGE and TLR4 Regulates HMGB1-Induced Inflammation by Promoting Cell Surface Expression of RAGE and TLR4. J. Immunol. 2020, 205, 767–775. [Google Scholar] [CrossRef]
- Zhu, L.; Ren, L.; Chen, Y.; Fang, J.; Ge, Z.; Li, X. Redox status of high-mobility group box 1 performs a dual role in angiogenesis of colorectal carcinoma. J. Cell. Mol. Med. 2015, 19, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Smith, D.R.; Tessler, L.; Mateo, A.R.; Martens, C.; Schartner, E.; Van der Ploeg, R.; Toth, C.; Zochodne, D.W.; Fernyhough, P. Receptor for advanced glycation end-products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp. Neurol. 2013, 249, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.S.; Li, J.; Liu, Y.; Wang, F.P.; Wang, S.Q.; She, W.M.; Wu, S.D.; Qi, X.L.; Zhou, Y.P.; Jiang, W. HMGB1-induced autophagy: A new pathway to maintain Treg function during chronic hepatitis B virus infection. Clin. Sci. 2017, 131, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Ling, Z.; Tan, X.; Chen, Y.; Huang, Q.; Li, M.; Liu, T.; Hou, C.; Chen, Y. High mobility group protein B1 (HMGB1) interacts with receptor for advanced glycation end products (RAGE) to promote airway smooth muscle cell proliferation through ERK and NF-kappaB pathways. Int. J. Clin. Exp. Pathol. 2019, 12, 3268–3278. [Google Scholar]
- Yamashiro, K.; Ideguchi, H.; Aoyagi, H.; Yoshihara-Hirata, C.; Hirai, A.; Suzuki-Kyoshima, R.; Zhang, Y.; Wake, H.; Nishibori, M.; Yamamoto, T.; et al. High Mobility Group Box 1 Expression in Oral Inflammation and Regeneration. Front. Immunol. 2020, 11, 1461. [Google Scholar] [CrossRef]
- Tomita, S.; Sekiguchi, F.; Kasanami, Y.; Naoe, K.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Cav3.2 overexpression in L4 dorsal root ganglion neurons after L5 spinal nerve cutting involves Egr-1, USP5 and HMGB1 in rats: An emerging signaling pathway for neuropathic pain. Eur. J. Pharmacol. 2020, 888, 173587. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef]
- Shah, B.S.; Burt, K.G.; Jacobsen, T.; Fernandes, T.D.; Alipui, D.O.; Weber, K.T.; Levine, M.; Chavan, S.S.; Yang, H.; Tracey, K.J.; et al. High mobility group box-1 induces pro-inflammatory signaling in human nucleus pulposus cells via toll-like receptor 4-dependent pathway. J. Orthop. Res. 2019, 37, 220–231. [Google Scholar] [CrossRef]
- Rudjito, R.; Agalave, N.M.; Farinotti, A.B.; Lundback, P.; Szabo-Pardi, T.; Price, T.J.; Harris, H.E.; Burton, M.D.; Svensson, C.I. Sex- and cell-dependent contribution of peripheral HMGB1 and TLR4 in arthritis-induced pain. Pain 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef]
- Mandke, P.; Vasquez, K.M. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: Implications in DNA repair and immune responses. DNA Repair. 2019, 83, 102701. [Google Scholar] [CrossRef] [PubMed]
- Nazari, A.; Khorramdelazad, H.; Hassanshahi, G. Biological/pathological functions of the CXCL12/CXCR4/CXCR7 axes in the pathogenesis of bladder cancer. Int. J. Clin. Oncol. 2017, 22, 991–1000. [Google Scholar] [CrossRef]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, X.; Xia, Z.; Chung, S.K.; Cheung, C.W. CXCL12/CXCR4 axis: An emerging neuromodulator in pathological pain. Rev. Neurosci. 2016, 27, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef]
- Okamoto, K.; Tamura, T.; Sawatsubashi, Y. Sepsis and disseminated intravascular coagulation. J. Intensive Care 2016, 4, 23. [Google Scholar] [CrossRef]
- Hayashi, Y.; Tsujita, R.; Tsubota, M.; Saeki, H.; Sekiguchi, F.; Honda, G.; Kawabata, A. Human soluble thrombomodulin-induced blockade of peripheral HMGB1-dependent allodynia in mice requires both the lectin-like and EGF-like domains. Biochem. Biophys. Res. Commun. 2018, 495, 634–638. [Google Scholar] [CrossRef]
- Chacur, M.; Milligan, E.D.; Gazda, L.S.; Armstrong, C.; Wang, H.; Tracey, K.J.; Maier, S.F.; Watkins, L.R. A new model of sciatic inflammatory neuritis (SIN): Induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain 2001, 94, 231–244. [Google Scholar] [CrossRef]
- Morioka, N.; Miyauchi, K.; Miyashita, K.; Kochi, T.; Zhang, F.F.; Nakamura, Y.; Liu, K.; Wake, H.; Hisaoka-Nakashima, K.; Nishibori, M.; et al. Spinal high-mobility group box-1 induces long-lasting mechanical hypersensitivity through the toll-like receptor 4 and upregulation of interleukin-1beta in activated astrocytes. J. Neurochem. 2019, 150, 738–758. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Sadanandan, J.; Chattopadhyay, M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2020, 21, 881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; de Carvalho-Barbosa, M.; Kavelaars, A.; Heijnen, C.J.; Albrecht, P.J.; Dougherty, P.M. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J. Pain 2016, 17, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Zhang, H.; Kosturakis, A.K.; Jawad, A.B.; Dougherty, P.M. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J. Pain 2014, 15, 712–725. [Google Scholar] [CrossRef]
- Luo, X.; Huh, Y.; Bang, S.; He, Q.; Zhang, L.; Matsuda, M.; Ji, R.R. Macrophage Toll-like Receptor 9 Contributes to Chemotherapy-Induced Neuropathic Pain in Male Mice. J. Neurosci. 2019, 39, 6848–6864. [Google Scholar] [CrossRef]
- Agalave, N.M.; Rudjito, R.; Farinotti, A.B.; Khoonsari, P.E.; Sandor, K.; Nomura, Y.; Szabo-Pardi, T.A.; Urbina, C.M.; Palada, V.; Price, T.J.; et al. Sex-dependent role of microglia in disulfide HMGB1-mediated mechanical hypersensitivity. Pain 2020. [Google Scholar] [CrossRef]
- Gu, H.; Wang, C.; Li, J.; Yang, Y.; Sun, W.; Jiang, C.; Li, Y.; Ni, M.; Liu, W.T.; Cheng, Z.; et al. High mobility group box-1-toll-like receptor 4-phosphatidylinositol 3-kinase/protein kinase B-mediated generation of matrix metalloproteinase-9 in the dorsal root ganglion promotes chemotherapy-induced peripheral neuropathy. Int. J. Cancer 2020, 146, 2810–2821. [Google Scholar] [CrossRef]
- Gao, Y.; Lv, X.; Yang, H.; Peng, L.; Ci, X. Isoliquiritigenin exerts antioxidative and anti-inflammatory effects via activating the KEAP-1/Nrf2 pathway and inhibiting the NF-kappaB and NLRP3 pathways in carrageenan-induced pleurisy. Food Funct. 2020, 11, 2522–2534. [Google Scholar] [CrossRef]
- Okamoto, T.; Tanigami, H.; Suzuki, K.; Shimaoka, M. Thrombomodulin: A bifunctional modulator of inflammation and coagulation in sepsis. Crit. Care Res. Pract. 2012, 2012, 614545. [Google Scholar] [CrossRef]
- Minami, T.; Takeda, M.; Sata, M.; Kato, H.; Yano, K.; Sakai, T.; Tsujita, R.; Kawasaki, K.; Ito, A. Thrombomodulin alfa prevents oxaliplatin-induced neuropathic symptoms through activation of thrombin-activatable fibrinolysis inhibitor and protein C without affecting anti-tumor activity. Eur. J. Pharmacol. 2020, 880, 173196. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Matsunawa, M.; Sano, F.; Miura, I. Efficacy of Recombinant Human Soluble Thrombomodulin in Treating Disseminated Intravascular Coagulation Complicating Allogeneic Hematopoietic Stem Cell Transplantation. Acta Haematol. 2018, 140, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339. [Google Scholar] [CrossRef]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, M.; Lambiase, A.; Armentano, M.; Tucciarone, G.; Bonfiglio, V.; Plateroti, R.; Alisi, L. The Complex Relationship between Diabetic Retinopathy and High-Mobility Group Box: A Review of Molecular Pathways and Therapeutic Strategies. Antioxidants 2020, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Peng, G.; Han, X. Glycyrrhizin ameliorates atopic dermatitis-like symptoms through inhibition of HMGB1. Int. Immunopharmacol. 2018, 60, 9–17. [Google Scholar] [CrossRef]
- Zhu, X.; Cong, J.; Lin, Z.; Sun, J.; Yang, B.; Li, A. Inhibition of HMGB1 Overcomes Resistance to Radiation and Chemotherapy in Nasopharyngeal Carcinoma. Onco Targets Ther. 2020, 13, 4189–4199. [Google Scholar] [CrossRef]
- Lei, X.; Hu, X.; Zhang, T.; Zhang, J.; Wu, C.; Hong, W.; Jiang, Y.; Wang, Q.; Xie, Y.; Zhao, Y.; et al. HMGB1 release promotes paclitaxel resistance in castration-resistant prostate cancer cells via activating c-Myc expression. Cell Signal. 2020, 72, 109631. [Google Scholar] [CrossRef]
- Sun, Y.; Hei, M.; Fang, Z.; Tang, Z.; Wang, B.; Hu, N. High-Mobility Group Box 1 Contributes to Cerebral Cortex Injury in a Neonatal Hypoxic-Ischemic Rat Model by Regulating the Phenotypic Polarization of Microglia. Front. Cell. Neurosci. 2019, 13, 506. [Google Scholar] [CrossRef]
- Le, K.; Wu, S.; Chibaatar, E.; Ali, A.I.; Guo, Y. Alarmin HMGB1 Plays a Detrimental Role in Hippocampal Dysfunction Caused by Hypoxia-Ischemia Insult in Neonatal Mice: Evidence from the Application of the HMGB1 Inhibitor Glycyrrhizin. ACS Chem. Neurosci. 2020, 11, 979–993. [Google Scholar] [CrossRef]
- Kuroiwa, Y.; Takakusagi, Y.; Kusayanagi, T.; Kuramochi, K.; Imai, T.; Hirayama, T.; Ito, I.; Yoshida, M.; Sakaguchi, K.; Sugawara, F. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS ONE 2013, 8, e63073. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Sakata, N.; Narumi, Y.; Kimura, T.; Hayashi, T.; Nagano, K.; Liu, K.; Nishibori, M.; Tsukita, S.; Yamada, T.; et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J. Biol. Chem. 2017, 292, 8436–8446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, X.; Cai, Y.; Yi, B.; Wen, Z. Metformin protects against hyperglycemia-induced cardiomyocytes injury by inhibiting the expressions of receptor for advanced glycation end products and high mobility group box 1 protein. Mol. Biol. Rep. 2014, 41, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yuan, F.; Deng, C.; He, F.; Zhang, Y.; Shen, H.; Chen, Z.; Qian, L. Metformin decreases LPS-induced inflammatory response in rabbit annulus fibrosus stem/progenitor cells by blocking HMGB1 release. Aging 2019, 11, 10252–10265. [Google Scholar] [CrossRef]
- Mao-Ying, Q.L.; Kavelaars, A.; Krukowski, K.; Huo, X.J.; Zhou, W.; Price, T.J.; Cleeland, C.; Heijnen, C.J. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS ONE 2014, 9, e100701. [Google Scholar] [CrossRef]
- Martinez, N.W.; Sanchez, A.; Diaz, P.; Broekhuizen, R.; Godoy, J.; Mondaca, S.; Catenaccio, A.; Macanas, P.; Nervi, B.; Calvo, M.; et al. Metformin protects from oxaliplatin induced peripheral neuropathy in rats. Neurobiol. Pain 2020, 8, 100048. [Google Scholar] [CrossRef]
- Riegsecker, S.; Wiczynski, D.; Kaplan, M.J.; Ahmed, S. Potential benefits of green tea polyphenol EGCG in the prevention and treatment of vascular inflammation in rheumatoid arthritis. Life Sci. 2013, 93, 307–312. [Google Scholar] [CrossRef]
- Li, W.; Zhu, S.; Li, J.; Assa, A.; Jundoria, A.; Xu, J.; Fan, S.; Eissa, N.T.; Tracey, K.J.; Sama, A.E.; et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem. Pharmacol. 2011, 81, 1152–1163. [Google Scholar] [CrossRef]
- Meng, X.Y.; Li, B.; Liu, S.; Kang, H.; Zhao, L.; Zhou, R. EGCG in Green Tea Induces Aggregation of HMGB1 Protein through Large Conformational Changes with Polarized Charge Redistribution. Sci. Rep. 2016, 6, 22128. [Google Scholar] [CrossRef]
- Choi, H.W.; Tian, M.; Song, F.; Venereau, E.; Preti, A.; Park, S.W.; Hamilton, K.; Swapna, G.V.; Manohar, M.; Moreau, M.; et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol. Med. 2015, 21, 526–535. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K.; et al. RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Mori, S.; Wake, H.; Zhang, J.; Liu, K.; Izushi, Y.; Takahashi, H.K.; Peng, B.; Nishibori, M. Establishment of in vitro binding assay of high mobility group box-1 and S100A12 to receptor for advanced glycation endproducts: Heparin’s effect on binding. Acta Med. Okayama 2009, 63, 203–211. [Google Scholar]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer. Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.; Bell, J.; Mancuso, J.Y.; Kupiec, J.W.; Sabbagh, M.N.; van Dyck, C.; Thomas, R.G.; Aisen, P.S.; Alzheimer’s Disease Cooperative, S. Clinical trial of an inhibitor of RAGE-Abeta interactions in Alzheimer disease. Neurology 2014, 82, 1536–1542. [Google Scholar] [CrossRef]
- Burstein, A.H.; Grimes, I.; Galasko, D.R.; Aisen, P.S.; Sabbagh, M.; Mjalli, A.M. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. 2014, 14, 12. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. mBio 2012, 3. [Google Scholar] [CrossRef]
- Cho, J.S.; Kang, J.H.; Um, J.Y.; Han, I.H.; Park, I.H.; Lee, H.M. Lipopolysaccharide induces pro-inflammatory cytokines and MMP production via TLR4 in nasal polyp-derived fibroblast and organ culture. PLoS ONE 2014, 9, e90683. [Google Scholar] [CrossRef]
- Abdelmageed, M.E.; El-Awady, M.S.; Abdelrahim, M.; Suddek, G.M. LPS-RS attenuation of lipopolysaccharide-induced acute lung injury involves NF-kappaB inhibition. Can. J. Physiol. Pharmacol. 2016, 94, 140–146. [Google Scholar] [CrossRef]
- Peri, F.; Calabrese, V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: An update. J. Med. Chem. 2014, 57, 3612–3622. [Google Scholar] [CrossRef]
- Rossignol, D.P.; Lynn, M. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J. Endotoxin Res. 2002, 8, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Cighetti, R.; Ciaramelli, C.; Sestito, S.E.; Zanoni, I.; Kubik, L.; Arda-Freire, A.; Calabrese, V.; Granucci, F.; Jerala, R.; Martin-Santamaria, S.; et al. Modulation of CD14 and TLR4.MD-2 activities by a synthetic lipid A mimetic. Chembiochem 2014, 15, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Aublin-Gex, A.; Sestito, S.E.; Shirey, K.A.; Patel, M.C.; Andre, P.; Blanco, J.C.; Vogel, S.N.; Peri, F.; Lotteau, V. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 2017, 7, 40791. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 2009, 157, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Ichikawa, T.; Ii, M.; Sunamoto, M.; Itoh, K.; Tamura, N.; Kitazaki, T. Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: Synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl)cyclohex-1-ene-1-carboxylate. J. Med. Chem. 2005, 48, 7457–7467. [Google Scholar] [CrossRef]
- Wang, J.; Tannous, B.A.; Poznansky, M.C.; Chen, H. CXCR4 antagonist AMD3100 (plerixafor): From an impurity to a therapeutic agent. Pharmacol. Res. 2020, 159, 105010. [Google Scholar] [CrossRef]
- Ulloa, L.; Ochani, M.; Yang, H.; Tanovic, M.; Halperin, D.; Yang, R.; Czura, C.J.; Fink, M.P.; Tracey, K.J. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc. Natl. Acad. Sci. USA 2002, 99, 12351–12356. [Google Scholar] [CrossRef]
- Simon, D.W.; Aneja, R.K.; Alexander, H.; Bell, M.J.; Bayir, H.; Kochanek, P.M.; Clark, R.S.B. Minocycline Attenuates High Mobility Group Box 1 Translocation, Microglial Activation, and Thalamic Neurodegeneration after Traumatic Brain Injury in Post-Natal Day 17 Rats. J. Neurotrauma. 2018, 35, 130–138. [Google Scholar] [CrossRef]
- Lohmann, K.L.; Vandenplas, M.L.; Barton, M.H.; Bryant, C.E.; Moore, J.N. The equine TLR4/MD-2 complex mediates recognition of lipopolysaccharide from Rhodobacter sphaeroides as an agonist. J. Endotoxin Res. 2007, 13, 235–242. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekiguchi, F.; Kawabata, A. Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2021, 22, 367. https://doi.org/10.3390/ijms22010367
Sekiguchi F, Kawabata A. Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy. International Journal of Molecular Sciences. 2021; 22(1):367. https://doi.org/10.3390/ijms22010367
Chicago/Turabian StyleSekiguchi, Fumiko, and Atsufumi Kawabata. 2021. "Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy" International Journal of Molecular Sciences 22, no. 1: 367. https://doi.org/10.3390/ijms22010367
APA StyleSekiguchi, F., & Kawabata, A. (2021). Role of HMGB1 in Chemotherapy-Induced Peripheral Neuropathy. International Journal of Molecular Sciences, 22(1), 367. https://doi.org/10.3390/ijms22010367