The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17

 and
and
Abstract
1. Introduction
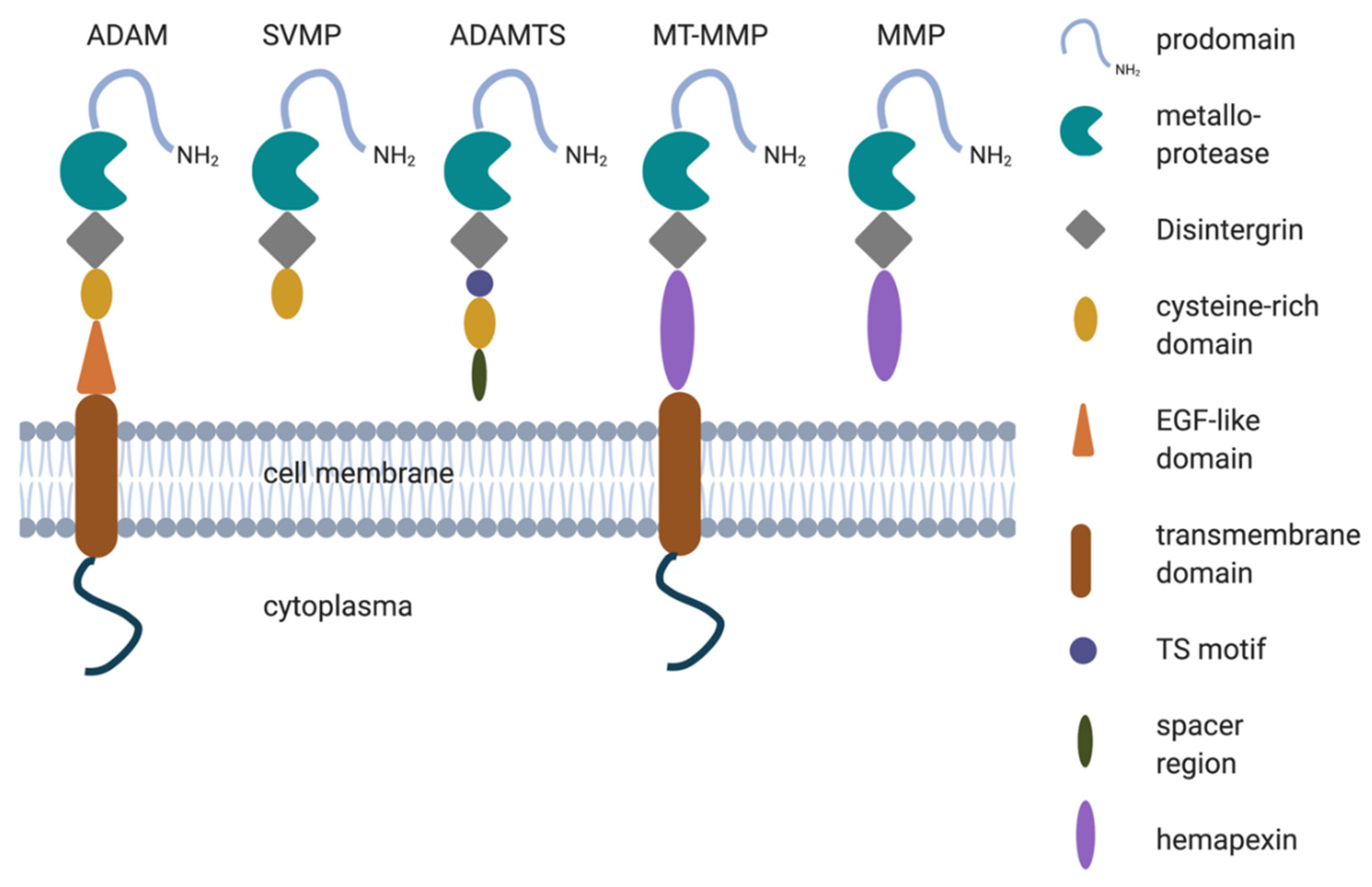
2. Structure of Metalloproteases
ADAMs
3. ADAM10 in the Central and Enteric Nervous Systems
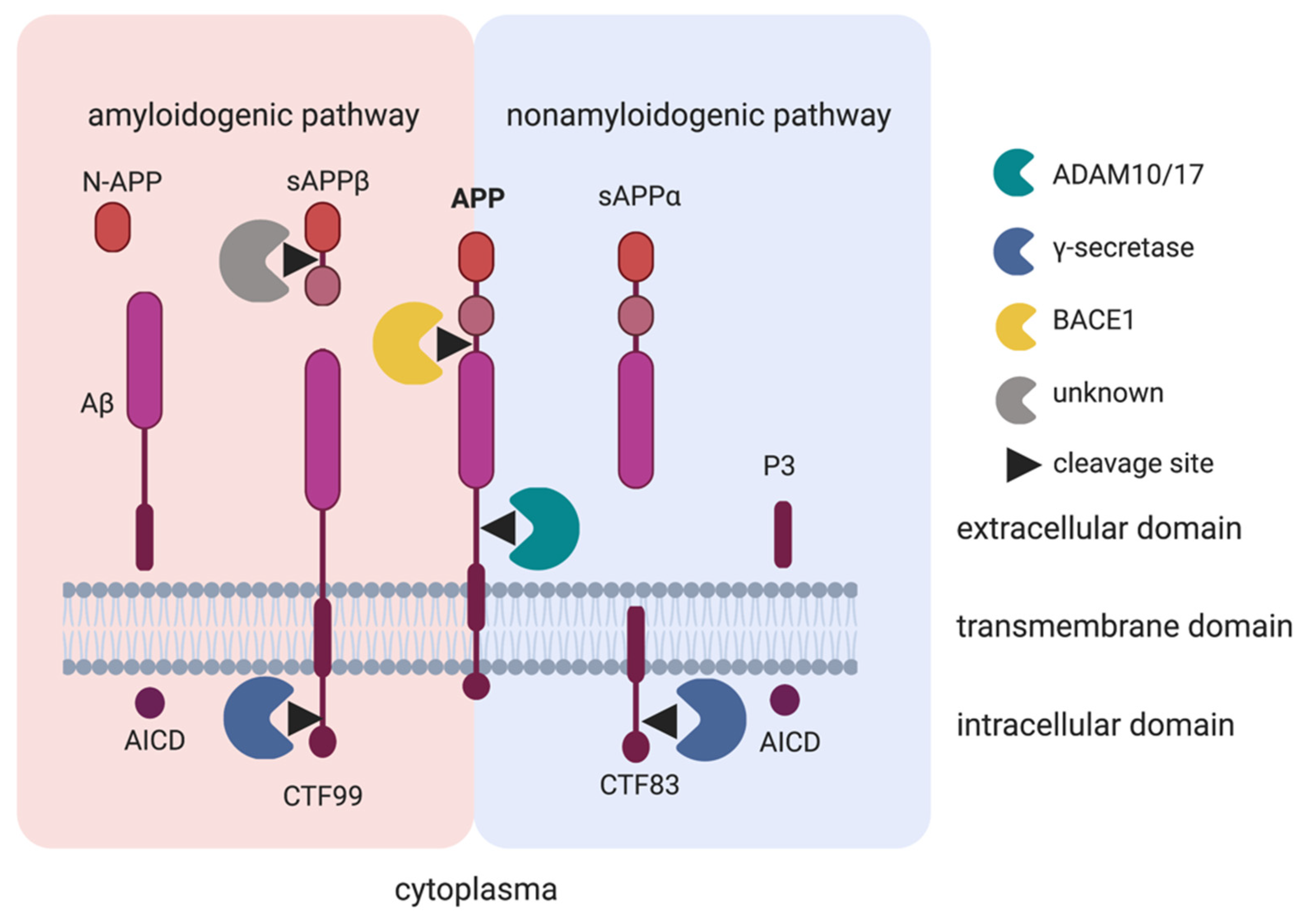
3.1. Amyloid-β Precursor Protein (APP)
3.2. Neuroligins (NLGNs) and Neurexins (NRXNs)
3.3. Protocadherins (PCDHs)
3.4. Neural Glial-Related Cell Adhesion Molecules (NrCAM)
3.5. Fractakine (CX3CL1)
4. ADAM17 in the Central Nervous System
4.1. Tumor Necrosis Factor-α (TNF-α)
4.2. Interleukin-6 Receptor (IL6-R)
4.3. Triggering Receptor Expressed in Myeloid Cells-2 (TREM2)
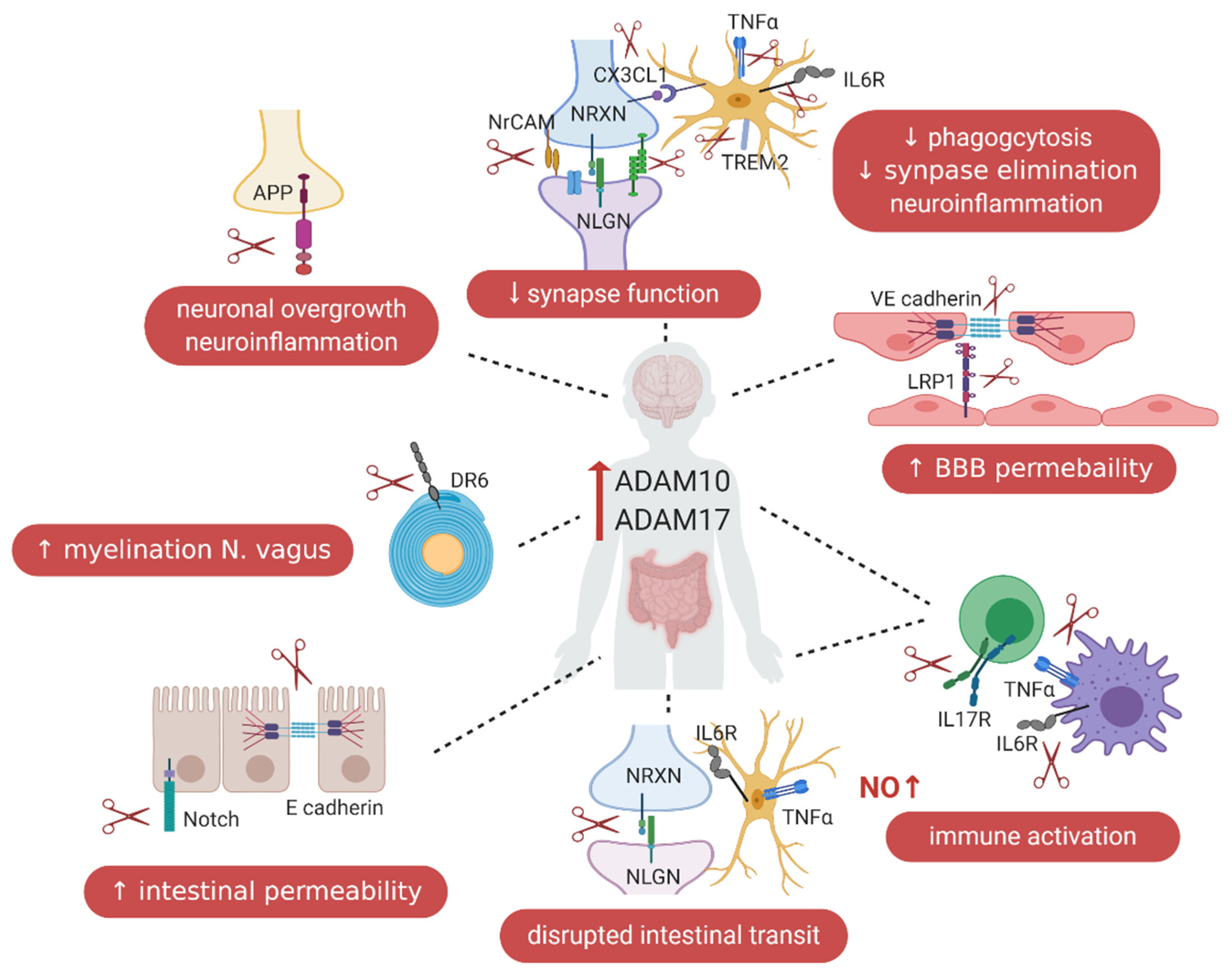
5. ADAM10 and ADAM17 in the Gut–Immune–Brain Axis
5.1. ADAM10 and ADAM17 and Blood–Brain Barrier Permeability
5.2. ADAM10 in the Intestinal Tract
5.3. ADAM17 in the Intestinal Tract
5.4. ADAMs and Intestinal Microbiota
5.5. ADAMs and the Immune System
6. Metalloproteases ADAM10 and ADAM17 as Therapeutic Targets for Autism Spectrum Disorders
6.1. TIMPs
6.2. ADAM Inhibitors
6.3. Probiotics, Bacterial Metabolites and Prebiotics?
6.4. Targeting ADAM10 and ADAM17 in ASD: Some Considerations
7. Outlook and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef]
- O’Reilly, C.; Lewis, J.D.; Elsabbagh, M. Is functional brain connectivity atypical in autism? A systematic review of EEG and MEG studies. PLoS ONE 2017, 12, e0175870. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef]
- Roussin, L.; Prince, N.; Perez-Pardo, P.; Kraneveld, A.D.; Rabot, S.; Naudon, L. Role of the Gut Microbiota in the Pathophysiology of Autism Spectrum Disorder: Clinical and Preclinical Evidence. Microorganisms 2020, 8, 1369. [Google Scholar] [CrossRef]
- DiStasio, M.M.; Nagakura, I.; Nadler, M.J.; Anderson, M.P. T lymphocytes and cytotoxic astrocyte blebs correlate across autism brains. Ann. Neurol. 2019, 86, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Glozier, N.; Dale, R.; Guastella, A.J. The Immune System, Cytokines, and Biomarkers in Autism Spectrum Disorder. Neurosci. Bull. 2017, 33, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Ormstad, H.; Bryn, V.; Saugstad, O.D.; Skjeldal, O.; Maes, M. Role of the Immune System in Autism Spectrum Disorders (ASD). CNS Neurol. Disord. Drug Targets 2018, 17, 489–495. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Dovgan, K.; Takahashi, N.; Beversdorf, D.Q. The Relationship among Gastrointestinal Symptoms, Problem Behaviors, and Internalizing Symptoms in Children and Adolescents with Autism Spectrum Disorder. Front. Psychiatry 2019, 10, 194. [Google Scholar] [CrossRef]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef]
- Ecker, C.; Bookheimer, S.Y.; Murphy, D.G. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015, 14, 1121–1134. [Google Scholar] [CrossRef]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Marui, T.; Funatogawa, I.; Koishi, S.; Yamamoto, K.; Matsumoto, H.; Hashimoto, O.; Nanba, E.; Nishida, H.; Sugiyama, T.; Kasai, K.; et al. Association of the neuronal cell adhesion molecule (NRCAM) gene variants with autism. Int. J. Neuropsychopharmacol. 2009, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Ray, B.; Long, J.M.; Sokol, D.K.; Lahiri, D.K. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: Proposal of a specific, anabolic pathway and putative biomarker. PLoS ONE 2011, 6, e20405. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Südhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef]
- Bruining, H.; Matsui, A.; Oguro-Ando, A.; Kahn, R.S.; Van’t Spijker, H.M.; Akkermans, G.; Stiedl, O.; van Engeland, H.; Koopmans, B.; van Lith, H.A.; et al. Genetic Mapping in Mice Reveals the Involvement of Pcdh9 in Long-Term Social and Object Recognition and Sensorimotor Development. Biol. Psychiatry 2015, 78, 485–495. [Google Scholar] [CrossRef]
- Moy, S.S.; Nonneman, R.J.; Young, N.B.; Demyanenko, G.P.; Maness, P.F. Impaired sociability and cognitive function in Nrcam-null mice. Behav. Brain Res. 2009, 205, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Bot, N.; Schweizer, C.; Ben Halima, S.; Fraering, P.C. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J. Biol. Chem. 2011, 286, 2762–2773. [Google Scholar] [CrossRef] [PubMed]
- Borcel, E.; Palczynska, M.; Krzisch, M.; Dimitrov, M.; Ulrich, G.; Toni, N.; Fraering, P.C. Shedding of neurexin 3β ectodomain by ADAM10 releases a soluble fragment that affects the development of newborn neurons. Sci. Rep. 2016, 6, 39310. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. eLife 2016, 5, e12748. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Muller, S.A.; Pan-Montojo, F.; Yoshida, F.; Fellgiebel, A.; Tomita, T.; Endres, K.; Lichtenthaler, S.F. NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol. Med. 2019, 11, e9695. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin proteases and their inhibitors: Foes or friends in nervous system physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar] [CrossRef] [PubMed]
- Huovila, A.P.; Turner, A.J.; Pelto-Huikko, M.; Kärkkäinen, I.; Ortiz, R.M. Shedding light on ADAM metalloproteinases. Trends Biochem. Sci. 2005, 30, 413–422. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Yu, X.; Wolslegel, K.; Nguyen, A.; Kung, C.; Chiang, E.; Kolumam, G.; Wei, N.; Wong, W.L.; DeForge, L.; et al. Lymphotoxin-alphabeta heterotrimers are cleaved by metalloproteinases and contribute to synovitis in rheumatoid arthritis. Cytokine 2010, 51, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Freour, T.; Jarry, A.; Bach-Ngohou, K.; Dejoie, T.; Bou-Hanna, C.; Denis, M.G.; Mosnier, J.F.; Laboisse, C.L.; Masson, D. TACE inhibition amplifies TNF-alpha-mediated colonic epithelial barrier disruption. Int. J. Mol. Med. 2009, 23, 41–48. [Google Scholar] [PubMed]
- Geesala, R.; Schanz, W.; Biggs, M.; Dixit, G.; Skurski, J.; Gurung, P.; Meyerholz, D.K.; Elliott, D.; Issuree, P.D.; Maretzky, T. Loss of RHBDF2 results in an early-onset spontaneous murine colitis. J. Leukoc. Biol. 2019, 105, 767–781. [Google Scholar] [CrossRef]
- Tsai, Y.H.; VanDussen, K.L.; Sawey, E.T.; Wade, A.W.; Kasper, C.; Rakshit, S.; Bhatt, R.G.; Stoeck, A.; Maillard, I.; Crawford, H.C.; et al. ADAM10 regulates Notch function in intestinal stem cells of mice. Gastroenterology 2014, 147, 822–834. [Google Scholar] [CrossRef]
- Ahmed, I.; Roy, B.C.; Raach, R.T.; Owens, S.M.; Xia, L.; Anant, S.; Sampath, V.; Umar, S. Enteric infection coupled with chronic Notch pathway inhibition alters colonic mucus composition leading to dysbiosis, barrier disruption and colitis. PLoS ONE 2018, 13, e0206701. [Google Scholar] [CrossRef]
- Dahan, S.; Rabinowitz, K.M.; Martin, A.P.; Berin, M.C.; Unkeless, J.C.; Mayer, L. Notch-1 signaling regulates intestinal epithelial barrier function, through interaction with CD4+ T cells, in mice and humans. Gastroenterology 2011, 140, 550–559. [Google Scholar] [CrossRef]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef]
- Lum, L.; Reid, M.S.; Blobel, C.P. Intracellular maturation of the mouse metalloprotease disintegrin MDC15. J. Biol. Chem. 1998, 273, 26236–26247. [Google Scholar] [CrossRef]
- Reiss, K.; Saftig, P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: Physiological and cellular functions. Semin. Cell Dev. Biol. 2009, 20, 126–137. [Google Scholar] [CrossRef]
- Fanjul-Fernández, M.; Folgueras, A.R.; Cabrera, S.; López-Otín, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.W.; Pearce, B.D.; Larsen, N.; Greaves-Lord, K.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Mortensen, E.L.; Grove, J. Amniotic fluid MMP-9 and neurotrophins in autism spectrum disorders: An exploratory study. Autism Res. 2012, 5, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 2016, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, W.; Grams, F.; Baumann, U.; Reinemer, P.; Gomis-Rüth, F.X.; McKay, D.B.; Bode, W. The metzincins--topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. Publ. Protein Soc. 1995, 4, 823–840. [Google Scholar] [CrossRef]
- Puente, X.S.; López-Otín, C. A genomic analysis of rat proteases and protease inhibitors. Genome Res. 2004, 14, 609–622. [Google Scholar] [CrossRef]
- Bode, W.; Gomis-Rüth, F.X.; Stöckler, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [CrossRef]
- Mochizuki, S.; Okada, Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007, 98, 621–628. [Google Scholar] [CrossRef]
- Düsterhöft, S.; Lokau, J.; Garbers, C. The metalloprotease ADAM17 in inflammation and cancer. Pathol. Res. Pract. 2019, 215, 152410. [Google Scholar] [CrossRef]
- Qian, M.; Shen, X.; Wang, H. The Distinct Role of ADAM17 in APP Proteolysis and Microglial Activation Related to Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 471–482. [Google Scholar] [CrossRef]
- Marcello, E.; Borroni, B.; Pelucchi, S.; Gardoni, F.; Di Luca, M. ADAM10 as a therapeutic target for brain diseases: From developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. [Google Scholar] [CrossRef]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M.G. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [PubMed]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bar, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell. Mol. Life Sci. 2018, 75, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.E.; Rubinstein, E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell. Mol. Life Sci. 2016, 73, 1895–1915. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.K.; Philips, M.A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
- Lundgren, J.L.; Ahmed, S.; Schedin-Weiss, S.; Gouras, G.K.; Winblad, B.; Tjernberg, L.O.; Frykman, S. ADAM10 and BACE1 are localized to synaptic vesicles. J. Neurochem. 2015, 135, 606–615. [Google Scholar] [CrossRef]
- Dempsey, P.J. Role of ADAM10 in intestinal crypt homeostasis and tumorigenesis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2228–2239. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993, 75, 1039–1042. [Google Scholar] [CrossRef]
- Sokol, D.K.; Chen, D.; Farlow, M.R.; Dunn, D.W.; Maloney, B.; Zimmer, J.A.; Lahiri, D.K. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J. Child Neurol. 2006, 21, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Westmark, C.J.; Westmark, P.R.; O’Riordan, K.J.; Ray, B.C.; Hervey, C.M.; Salamat, M.S.; Abozeid, S.H.; Stein, K.M.; Stodola, L.A.; Tranfaglia, M.; et al. Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS ONE 2011, 6, e26549. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Sokol, D.K.; Erickson, C.; Ray, B.; Ho, C.Y.; Maloney, B. Autism as early neurodevelopmental disorder: Evidence for an sAPPα-mediated anabolic pathway. Front. Cell. Neurosci. 2013, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, C.; Beyaert, R.; van Loo, G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011, 34, 619–628. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Hu, Y.; Ji, B.; Shao, Z.; Yang, W.; Huang, G.; Walus, L.; Rhodes, K.; Gong, B.J.; et al. Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat. Med. 2011, 17, 816–821. [Google Scholar] [CrossRef]
- Popko, B. Downregulating DR6 to drive remyelination. Nat. Med. 2011, 17, 779–780. [Google Scholar] [CrossRef]
- Colombo, A.; Hsia, H.E.; Wang, M.; Kuhn, P.H.; Brill, M.S.; Canevazzi, P.; Feederle, R.; Taveggia, C.; Misgeld, T.; Lichtenthaler, S.F. Non-cell-autonomous function of DR6 in Schwann cell proliferation. EMBO J. 2018, 37, e97390. [Google Scholar] [CrossRef]
- Corfas, G.; Velardez, M.O.; Ko, C.P.; Ratner, N.; Peles, E. Mechanisms and roles of axon-Schwann cell interactions. J. Neurosci. 2004, 24, 9250–9260. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Medina, I.; Jevans, B.; Boismoreau, F.; Chettouh, Z.; Enomoto, H.; Muller, T.; Birchmeier, C.; Burns, A.J.; Brunet, J.F. Dual origin of enteric neurons in vagal Schwann cell precursors and the sympathetic neural crest. Proc. Natl. Acad. Sci. USA 2017, 114, 11980–11985. [Google Scholar] [CrossRef] [PubMed]
- Vega, C.; Martiel, J.L.; Drouhault, D.; Burckhart, M.F.; Coles, J.A. Uptake of locally applied deoxyglucose, glucose and lactate by axons and Schwann cells of rat vagus nerve. J. Physiol. 2003, 546, 551–564. [Google Scholar] [CrossRef]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259. [Google Scholar] [CrossRef] [PubMed]
- van Hoorn, A.; Carpenter, T.; Oak, K.; Laugharne, R.; Ring, H.; Shankar, R. Neuromodulation of autism spectrum disorders using vagal nerve stimulation. J. Clin. Neurosci. 2019, 63, 8–12. [Google Scholar] [CrossRef]
- Jin, Y.; Kong, J. Transcutaneous Vagus Nerve Stimulation: A Promising Method for Treatment of Autism Spectrum Disorders. Front. Neurosci. 2016, 10, 609. [Google Scholar] [CrossRef]
- Krueger, D.D.; Tuffy, L.P.; Papadopoulos, T.; Brose, N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 2012, 22, 412–422. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 213. [Google Scholar] [CrossRef]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Budreck, E.C.; Scheiffele, P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur. J. Neurosci. 2007, 26, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hayashi, Y.; Nakahara, S.; Kumazaki, H.; Prox, J.; Horiuchi, K.; Zeng, M.; Tanimura, S.; Nishiyama, Y.; Osawa, S.; et al. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 2012, 76, 410–422. [Google Scholar] [CrossRef]
- Trotter, J.H.; Hao, J.; Maxeiner, S.; Tsetsenis, T.; Liu, Z.; Zhuang, X.; Südhof, T.C. Synaptic neurexin-1 assembles into dynamically regulated active zone nanoclusters. J. Cell Biol. 2019, 218, 2677–2698. [Google Scholar] [CrossRef] [PubMed]
- Hosie, S.; Ellis, M.; Swaminathan, M.; Ramalhosa, F.; Seger, G.O.; Balasuriya, G.K.; Gillberg, C.; Råstam, M.; Churilov, L.; McKeown, S.J.; et al. Gastrointestinal dysfunction in patients and mice expressing the autism-associated R451C mutation in neuroligin-3. Autism Res. 2019, 12, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Sharna, S.S.; Balasuriya, G.K.; Hosie, S.; Nithianantharajah, J.; Franks, A.E.; Hill-Yardin, E.L. Altered Caecal Neuroimmune Interactions in the Neuroligin-3(R451C) Mouse Model of Autism. Front. Cell. Neurosci. 2020, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Leembruggen, A.J.L.; Balasuriya, G.K.; Zhang, J.; Schokman, S.; Swiderski, K.; Bornstein, J.C.; Nithianantharajah, J.; Hill-Yardin, E.L. Colonic dilation and altered ex vivo gastrointestinal motility in the neuroligin-3 knockout mouse. Autism Res. 2020, 13, 691–701. [Google Scholar] [CrossRef]
- Gabriele, S.; Sacco, R.; Altieri, L.; Neri, C.; Urbani, A.; Bravaccio, C.; Riccio, M.P.; Iovene, M.R.; Bombace, F.; De Magistris, L.; et al. Slow intestinal transit contributes to elevate urinary p-cresol level in Italian autistic children. Autism Res. 2016, 9, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.J.; Langefeld, C.D.; Zimmerman, K.; Schwartz, M.Z.; Krigsman, A. A molecular biomarker for prediction of clinical outcome in children with ASD, constipation, and intestinal inflammation. Sci. Rep. 2019, 9, 5987. [Google Scholar] [CrossRef]
- Wang, D.; Pan, J.; Song, G.; Gao, N.; Zheng, Y.; Zhang, Q.; Li, A. Abundance and Significance of Neuroligin-1 and Neurexin II in the Enteric Nervous System of Embryonic Rats. BioMed Res. Int. 2017, 2017, 1209360. [Google Scholar] [CrossRef]
- Horder, J.; Petrinovic, M.M.; Mendez, M.A.; Bruns, A.; Takumi, T.; Spooren, W.; Barker, G.J.; Künnecke, B.; Murphy, D.G. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl. Psychiatry 2018, 8, 106. [Google Scholar] [CrossRef]
- Peixoto, R.T.; Kunz, P.A.; Kwon, H.; Mabb, A.M.; Sabatini, B.L.; Philpot, B.D.; Ehlers, M.D. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 2012, 76, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Ellegood, J.; Lerch, J.P.; Henkelman, R.M. Brain abnormalities in a Neuroligin3 R451C knockin mouse model associated with autism. Autism Res. 2011, 4, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Halbleib, J.M.; Nelson, W.J. Cadherins in development: Cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006, 20, 3199–3214. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Tanihara, H.; Heimark, R.L.; Obata, S.; Davidson, M.; St John, T.; Taketani, S.; Suzuki, S. Protocadherins: A large family of cadherin-related molecules in central nervous system. EMBO J. 1993, 12, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Tanaka, H.; Sugiura, H.; Okamura, K.; Sakaguchi, T.; Tran, U.; Takemiya, T.; Mizoguchi, A.; Yagita, Y.; Sakurai, T.; et al. Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron 2007, 56, 456–471. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Rafi, S.K.; Hossain, W.; Stephan, D.A.; Manzardo, A.M. Whole exome sequencing in females with autism implicates novel and candidate genes. Int. J. Mol. Sci. 2015, 16, 1312–1335. [Google Scholar] [CrossRef] [PubMed]
- Breuillard, D.; Leunen, D.; Chemaly, N.; Auclair, L.; Pinard, J.M.; Kaminska, A.; Desguerre, I.; Ouss, L.; Nabbout, R. Autism spectrum disorder phenotype and intellectual disability in females with epilepsy and PCDH-19 mutations. Epilepsy Behav. 2016, 60, 75–80. [Google Scholar] [CrossRef]
- Grumet, M.; Mauro, V.; Burgoon, M.P.; Edelman, G.M.; Cunningham, B.A. Structure of a new nervous system glycoprotein, Nr-CAM, and its relationship to subgroups of neural cell adhesion molecules. J. Cell Biol. 1991, 113, 1399–1412. [Google Scholar] [CrossRef]
- Zelina, P.; Avci, H.X.; Thelen, K.; Pollerberg, G.E. The cell adhesion molecule NrCAM is crucial for growth cone behaviour and pathfinding of retinal ganglion cell axons. Development 2005, 132, 3609–3618. [Google Scholar] [CrossRef]
- Demyanenko, G.P.; Mohan, V.; Zhang, X.; Brennaman, L.H.; Dharbal, K.E.; Tran, T.S.; Manis, P.B.; Maness, P.F. Neural cell adhesion molecule NrCAM regulates Semaphorin 3F-induced dendritic spine remodeling. J. Neurosci. 2014, 34, 11274–11287. [Google Scholar] [CrossRef]
- Sakurai, T.; Lustig, M.; Nativ, M.; Hemperly, J.J.; Schlessinger, J.; Peles, E.; Grumet, M. Induction of neurite outgrowth through contactin and Nr-CAM by extracellular regions of glial receptor tyrosine phosphatase beta. J. Cell Biol. 1997, 136, 907–918. [Google Scholar] [CrossRef]
- Prox, J.; Bernreuther, C.; Altmeppen, H.; Grendel, J.; Glatzel, M.; D’Hooge, R.; Stroobants, S.; Ahmed, T.; Balschun, D.; Willem, M.; et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 2013, 33, 12915–12928. [Google Scholar] [CrossRef]
- Mohan, V.; Sullivan, C.S.; Guo, J.; Wade, S.D.; Majumder, S.; Agarwal, A.; Anton, E.S.; Temple, B.S.; Maness, P.F. Temporal Regulation of Dendritic Spines Through NrCAM-Semaphorin3F Receptor Signaling in Developing Cortical Pyramidal Neurons. Cereb. Cortex 2019, 29, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Heyden, A.; Angenstein, F.; Sallaz, M.; Seidenbecher, C.; Montag, D. Abnormal axonal guidance and brain anatomy in mouse mutants for the cell recognition molecules close homolog of L1 and NgCAM-related cell adhesion molecule. Neuroscience 2008, 155, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Lamb, J.A.; Barnby, G.; Sykes, N.; Moberly, T.; Beyer, K.S.; Klauck, S.M.; Poustka, F.; Bacchelli, E.; Blasi, F.; et al. Mutation screening and association analysis of six candidate genes for autism on chromosome 7q. Eur. J. Hum. Genet. 2005, 13, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, H.B.; Olson, L.M.; Bradford, Y.; Folstein, S.E.; Santangelo, S.L.; Sutcliffe, J.S.; Haines, J.L. Examination of NRCAM, LRRN3, KIAA0716, and LAMB1 as autism candidate genes. BMC Med. Genet. 2004, 5, 12. [Google Scholar] [CrossRef]
- Rostene, W.; Kitabgi, P.; Parsadaniantz, S.M. Chemokines: A new class of neuromodulator? Nat. Rev. Neurosci. 2007, 8, 895–903. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- O’Sullivan, S.A.; Gasparini, F.; Mir, A.K.; Dev, K.K. Fractalkine shedding is mediated by p38 and the ADAM10 protease under pro-inflammatory conditions in human astrocytes. J. Neuroinflamm. 2016, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, M.D.; Krull, J.E.; Douglass, J.D.; Fasnacht, R.; Lara-Lince, F.; Meek, T.H.; Shi, X.; Damian, V.; Nguyen, H.T.; Matsen, M.E.; et al. Sex differences in microglial CX3CR1 signalling determine obesity susceptibility in mice. Nat. Commun. 2017, 8, 14556. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef]
- Ray, B.; Sokol, D.K.; Maloney, B.; Lahiri, D.K. Finding novel distinctions between the sAPPα-mediated anabolic biochemical pathways in Autism Spectrum Disorder and Fragile X Syndrome plasma and brain tissue. Sci. Rep. 2016, 6, 26052. [Google Scholar] [CrossRef]
- Bell, J.H.; Herrera, A.H.; Li, Y.; Walcheck, B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 2007, 82, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Mezyk-Kopec, R.; Bzowska, M.; Stalinska, K.; Chelmicki, T.; Podkalicki, M.; Jucha, J.; Kowalczyk, K.; Mak, P.; Bereta, J. Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 2009, 46, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Lucchina, L.; Depino, A.M. Altered peripheral and central inflammatory responses in a mouse model of autism. Autism Res. 2014, 7, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- Garbers, C.; Janner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Dusterhoft, S.; Zhu, Y.; Grotzinger, J.; et al. Proteolytic Origin of the Soluble Human IL-6R In Vivo and a Decisive Role of N-Glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef]
- Wei, H.; Chadman, K.K.; McCloskey, D.P.; Sheikh, A.M.; Malik, M.; Brown, W.T.; Li, X. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim. Biophys. Acta 2012, 1822, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ma, Y.; Liu, J.; Ding, C.; Jin, G.; Wang, Y.; Hu, F.; Yu, L. Inhibition of IL-6 trans-signaling in the brain increases sociability in the BTBR mouse model of autism. Biochim. Biophys. Acta 2016, 1862, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Feuerbach, D.; Schindler, P.; Barske, C.; Joller, S.; Beng-Louka, E.; Worringer, K.A.; Kommineni, S.; Kaykas, A.; Ho, D.J.; Ye, C.; et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci. Lett. 2017, 660, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef]
- Holingue, C.; Newill, C.; Lee, L.C.; Pasricha, P.J.; Daniele Fallin, M. Gastrointestinal symptoms in autism spectrum disorder: A review of the literature on ascertainment and prevalence. Autism Res. 2018, 11, 24–36. [Google Scholar] [CrossRef]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Perez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal symptoms in autism spectrum disorder: A meta-analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef]
- Lasheras, I.; Seral, P.; Latorre, E.; Barroso, E.; Gracia-Garcia, P.; Santabarbara, J. Microbiota and gut-brain axis dysfunction in autism spectrum disorder: Evidence for functional gastrointestinal disorders. Asian J. Psychiatry 2020, 47, 101874. [Google Scholar] [CrossRef] [PubMed]
- Fowlie, G.; Cohen, N.; Ming, X. The Perturbance of Microbiome and Gut-Brain Axis in Autism Spectrum Disorders. Int. J. Mol. Sci. 2018, 19, 2251. [Google Scholar] [CrossRef] [PubMed]
- de Magistris, L.; Familiari, V.; Pascotto, A.; Sapone, A.; Frolli, A.; Iardino, P.; Carteni, M.; De Rosa, M.; Francavilla, R.; Riegler, G.; et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J. Pediatric Gastroenterol. Nutr. 2010, 51, 418–424. [Google Scholar] [CrossRef]
- Liu, Z.; Li, N.; Neu, J. Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr. 2005, 94, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav. Immun. 2015, 44, 1–8. [Google Scholar] [CrossRef]
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 2012, 19, 121–130. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef]
- Candela, P.; Saint-Pol, J.; Kuntz, M.; Boucau, M.C.; Lamartiniere, Y.; Gosselet, F.; Fenart, L. In vitro discrimination of the role of LRP1 at the BBB cellular level: Focus on brain capillary endothelial cells and brain pericytes. Brain Res. 2015, 1594, 15–26. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef]
- Shackleton, B.; Crawford, F.; Bachmeier, C. Inhibition of ADAM10 promotes the clearance of Aβ across the BBB by reducing LRP1 ectodomain shedding. Fluids Barriers CNS 2016, 13, 14. [Google Scholar] [CrossRef]
- Liang, S.J.; Li, X.G.; Wang, X.Q. Notch Signaling in Mammalian Intestinal Stem Cells: Determining Cell Fate and Maintaining Homeostasis. Curr. Stem Cell Res. Ther. 2019, 14, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, K.K.; Orsso, C.E.; Richard, C.; Haqq, A.M.; Zwaigenbaum, L. Risk Factors for Unhealthy Weight Gain and Obesity among Children with Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 3285. [Google Scholar] [CrossRef] [PubMed]
- Puig, K.L.; Brose, S.A.; Zhou, X.; Sens, M.A.; Combs, G.F.; Jensen, M.D.; Golovko, M.Y.; Combs, C.K. Amyloid precursor protein modulates macrophage phenotype and diet-dependent weight gain. Sci. Rep. 2017, 7, 43725. [Google Scholar] [CrossRef]
- Puig, K.L.; Manocha, G.D.; Combs, C.K. Amyloid precursor protein mediated changes in intestinal epithelial phenotype in vitro. PLoS ONE 2015, 10, e0119534. [Google Scholar] [CrossRef] [PubMed]
- Puig, K.L.; Swigost, A.J.; Zhou, X.; Sens, M.A.; Combs, C.K. Amyloid precursor protein expression modulates intestine immune phenotype. J. Neuroimmune Pharmacol. 2012, 7, 215–230. [Google Scholar] [CrossRef] [PubMed]
- An, Y.A.; Crewe, C.; Asterholm, I.W.; Sun, K.; Chen, S.; Zhang, F.; Shao, M.; Funcke, J.B.; Zhang, Z.; Straub, L.; et al. Dysregulation of Amyloid Precursor Protein Impairs Adipose Tissue Mitochondrial Function and Promotes Obesity. Nat. Metab. 2019, 1, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef]
- Wang, F.; Graham, W.V.; Wang, Y.; Witkowski, E.D.; Schwarz, B.T.; Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005, 166, 409–419. [Google Scholar] [CrossRef]
- Moriez, R.; Salvador-Cartier, C.; Theodorou, V.; Fioramonti, J.; Eutamene, H.; Bueno, L. Myosin light chain kinase is involved in lipopolysaccharide-induced disruption of colonic epithelial barrier and bacterial translocation in rats. Am. J. Pathol. 2005, 167, 1071–1079. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Bambini-Junior, V.; Zanatta, G.; Della Flora Nunes, G.; Mueller de Melo, G.; Michels, M.; Fontes-Dutra, M.; Nogueira Freire, V.; Riesgo, R.; Gottfried, C. Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci. Lett. 2014, 583, 176–181. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Alzahrani, M.Z.; Alshammari, M.A.; Alanazi, W.A.; Alasmari, A.F.; Attia, S.M. Resveratrol attenuates pro-inflammatory cytokines and activation of JAK1-STAT3 in BTBR T(+) Itpr3(tf)/J autistic mice. Eur. J. Pharmacol. 2018, 829, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol. Psychiatry 2015, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef]
- Coretti, L.; Paparo, L.; Riccio, M.P.; Amato, F.; Cuomo, M.; Natale, A.; Borrelli, L.; Corrado, G.; Comegna, M.; Buommino, E.; et al. Gut Microbiota Features in Young Children With Autism Spectrum Disorders. Front. Microbiol. 2018, 9, 3146. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E.; Orsi, P.; Boso, M.; Broglia, D.; Brondino, N.; Barale, F.; di Nemi, S.U.; Politi, P. Low-grade endotoxemia in patients with severe autism. Neurosci. Lett. 2010, 471, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Muñoz, I.; Compte, M.; Álvarez-Cienfuegos, A.; Álvarez-Vallina, L.; Sanz, L. Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Yao, L.; Kan, E.M.; Lu, J.; Hao, A.; Dheen, S.T.; Kaur, C.; Ling, E.A. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: Role of TLR4 in hypoxic microglia. J. Neuroinflamm. 2013, 10, 23. [Google Scholar] [CrossRef]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef]
- Maretzky, T.; Scholz, F.; Köten, B.; Proksch, E.; Saftig, P.; Reiss, K. ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Investig. Dermatol. 2008, 128, 1737–1746. [Google Scholar] [CrossRef]
- Alshammari, M.K.; AlKhulaifi, M.M.; Al Farraj, D.A.; Somily, A.M.; Albarrag, A.M. Incidence of Clostridium perfringens and its toxin genes in the gut of children with autism spectrum disorder. Anaerobe 2020, 61, 102114. [Google Scholar] [CrossRef]
- Gora, B.; Gofron, Z.; Grosiak, M.; Aptekorz, M.; Kazek, B.; Kocelak, P.; Radosz-Komoniewska, H.; Chudek, J.; Martirosian, G. Toxin profile of fecal Clostridium perfringens strains isolated from children with autism spectrum disorders. Anaerobe 2018, 51, 73–77. [Google Scholar] [CrossRef]
- Popoff, M.R.; Bouvet, P. Clostridial toxins. Future Microbiol. 2009, 4, 1021–1064. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Takehara, M.; Kobayashi, K.; Nagahama, M. Clostridium perfringens Delta-Toxin Damages the Mouse Small Intestine. Toxins 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Seike, S.; Takehara, M.; Takagishi, T.; Miyamoto, K.; Kobayashi, K.; Nagahama, M. Delta-toxin from Clostridium perfringens perturbs intestinal epithelial barrier function in Caco-2 cell monolayers. Biochim. Biophys. Acta Biomembr. 2018, 1860, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Napolioni, V. Urinary p-cresol in autism spectrum disorder. Neurotoxicol. Teratol. 2013, 36, 82–90. [Google Scholar] [CrossRef]
- Altieri, L.; Neri, C.; Sacco, R.; Curatolo, P.; Benvenuto, A.; Muratori, F.; Santocchi, E.; Bravaccio, C.; Lenti, C.; Saccani, M.; et al. Urinary p-cresol is elevated in small children with severe autism spectrum disorder. Biomarkers 2011, 16, 252–260. [Google Scholar] [CrossRef]
- Mishra, H.K.; Johnson, T.J.; Seelig, D.M.; Walcheck, B. Targeting ADAM17 in leukocytes increases neutrophil recruitment and reduces bacterial spread during polymicrobial sepsis. J. Leukoc. Biol. 2016, 100, 999–1004. [Google Scholar] [CrossRef]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical Relevance and Role of Neuronal AT(1) Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Chodavarapu, H.; Porretta, C.; Robinson, L.K.; Lazartigues, E. Dynamics of ADAM17-Mediated Shedding of ACE2 Applied to Pancreatic Islets of Male db/db Mice. Endocrinology 2015, 156, 4411–4425. [Google Scholar] [CrossRef]
- Cole-Jeffrey, C.T.; Liu, M.; Katovich, M.J.; Raizada, M.K.; Shenoy, V. ACE2 and Microbiota: Emerging Targets for Cardiopulmonary Disease Therapy. J. Cardiovasc. Pharmacol. 2015, 66, 540–550. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Neidhöfer, S.; Matthias, T. The Gut Microbiome Feelings of the Brain: A Perspective for Non-Microbiologists. Microorganisms 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef]
- Cerwenka, A.; Swain, S.L. TGF-beta1: Immunosuppressant and viability factor for T lymphocytes. Microbes Infect. 1999, 1, 1291–1296. [Google Scholar] [CrossRef]
- Ashwood, P.; Enstrom, A.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.L.; Croen, L.A.; Ozonoff, S.; Pessah, I.N.; Van de Water, J. Decreased transforming growth factor beta1 in autism: A potential link between immune dysregulation and impairment in clinical behavioral outcomes. J. Neuroimmunol. 2008, 204, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Hashimoto, K.; Iwata, Y.; Nakamura, K.; Tsujii, M.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Decreased serum levels of transforming growth factor-beta1 in patients with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Al-Ayadhi, L.; Alhowikan, A.M.; Halepoto, D.M. Impact of Auditory Integrative Training on Transforming Growth Factor-β1 and Its Effect on Behavioral and Social Emotions in Children with Autism Spectrum Disorder. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2018, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Chang, C.F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Ke, K.F.; Lu, J.H.; Qiu, Y.H.; Peng, Y.P. Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer’s disease model rats. PLoS ONE 2015, 10, e0116549. [Google Scholar] [CrossRef]
- Depino, A.M.; Lucchina, L.; Pitossi, F. Early and adult hippocampal TGF-β1 overexpression have opposite effects on behavior. Brain Behav. Immun. 2011, 25, 1582–1591. [Google Scholar] [CrossRef]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFβ signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef]
- Liu, C.; Xu, P.; Lamouille, S.; Xu, J.; Derynck, R. TACE-mediated ectodomain shedding of the type I TGF-beta receptor downregulates TGF-beta signaling. Mol. Cell 2009, 35, 26–36. [Google Scholar] [CrossRef]
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFβ signaling through the cleavage of vasorin. Oncogene 2011, 30, 1912–1922. [Google Scholar] [CrossRef]
- Ikeda, Y.; Imai, Y.; Kumagai, H.; Nosaka, T.; Morikawa, Y.; Hisaoka, T.; Manabe, I.; Maemura, K.; Nakaoka, T.; Imamura, T.; et al. Vasorin, a transforming growth factor beta-binding protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 10732–10737. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.M.; Bishop, K.; Fox, D.; Zou, W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011, 32, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Kuchroo, V.K.; Awasthi, A. Emerging new roles of Th17 cells. Eur. J. Immunol. 2012, 42, 2211–2214. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Yan, I.; Schwarz, J.; Lücke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.; Rose-John, S.; et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J. Leukoc. Biol. 2016, 99, 749–760. [Google Scholar] [CrossRef]
- Liu, S.; Fu, Y.; Mei, K.; Jiang, Y.; Sun, X.; Wang, Y.; Ren, F.; Jiang, C.; Meng, L.; Lu, S.; et al. A shedding soluble form of interleukin-17 receptor D exacerbates collagen-induced arthritis through facilitating TNF-α-dependent receptor clustering. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J. Cell Sci. 2002, 115, 3719–3727. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Amour, A.; Knight, C.G.; Webster, A.; Slocombe, P.M.; Stephens, P.E.; Knäuper, V.; Docherty, A.J.; Murphy, G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000, 473, 275–279. [Google Scholar] [CrossRef]
- Amour, A.; Slocombe, P.M.; Webster, A.; Butler, M.; Knight, C.G.; Smith, B.J.; Stephens, P.E.; Shelley, C.; Hutton, M.; Knäuper, V.; et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998, 435, 39–44. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 44, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, G.; Menon, V.; Jugdutt, B.I. Improved balance between TIMP-3 and MMP-9 after regional myocardial ischemia-reperfusion during AT1 receptor blockade. J. Card. Fail. 2004, 10, 442–449. [Google Scholar] [CrossRef]
- Hoettecke, N.; Ludwig, A.; Foro, S.; Schmidt, B. Improved synthesis of ADAM10 inhibitor GI254023X. Neuro-Degener. Dis. 2010, 7, 232–238. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Mahasenan, K.V.; Ding, D.; Gao, M.; Nguyen, T.T.; Suckow, M.A.; Schroeder, V.A.; Wolter, W.R.; Chang, M.; Mobashery, S. In Search of Selectivity in Inhibition of ADAM10. ACS Med. Chem. Lett. 2018, 9, 708–713. [Google Scholar] [CrossRef]
- Hirata, S.; Murata, T.; Suzuki, D.; Nakamura, S.; Jono-Ohnishi, R.; Hirose, H.; Sawaguchi, A.; Nishimura, S.; Sugimoto, N.; Eto, K. Selective Inhibition of ADAM17 Efficiently Mediates Glycoprotein Ibα Retention During Ex Vivo Generation of Human Induced Pluripotent Stem Cell-Derived Platelets. Stem Cells Transl. Med. 2017, 6, 720–730. [Google Scholar] [CrossRef]
- Abdellatif, B.; McVeigh, C.; Bendriss, G.; Chaari, A. The Promising Role of Probiotics in Managing the Altered Gut in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 4159. [Google Scholar] [CrossRef]
- Navarro, F.; Liu, Y.; Rhoads, J.M. Can probiotics benefit children with autism spectrum disorders? World J. Gastroenterol. 2016, 22, 10093–10102. [Google Scholar] [CrossRef]
- Liu, J.; Wan, G.B.; Huang, M.S.; Agyapong, G.; Zou, T.L.; Zhang, X.Y.; Liu, Y.W.; Song, Y.Q.; Tsai, Y.C.; Kong, X.J. Probiotic Therapy for Treating Behavioral and Gastrointestinal Symptoms in Autism Spectrum Disorder: A Systematic Review of Clinical Trials. Curr. Med. Sci. 2019, 39, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, R.; Gibson, G.R.; Vulevic, J.; Giallourou, N.; Castro-Mejía, J.L.; Hansen, L.H.; Leigh Gibson, E.; Nielsen, D.S.; Costabile, A. A prebiotic intervention study in children with autism spectrum disorders (ASDs). Microbiome 2018, 6, 133. [Google Scholar] [CrossRef]
- Carlezon, W.A., Jr.; Kim, W.; Missig, G.; Finger, B.C.; Landino, S.M.; Alexander, A.J.; Mokler, E.L.; Robbins, J.O.; Li, Y.; Bolshakov, V.Y.; et al. Maternal and early postnatal immune activation produce sex-specific effects on autism-like behaviors and neuroimmune function in mice. Sci. Rep. 2019, 9, 16928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, F.; Xie, Y.; Lin, Y.; Zhao, T.; Tao, S.; Lai, Z.; Wei, N.; Yang, R.; Shao, Y.; et al. miR-23b Negatively Regulates Sepsis-Induced Inflammatory Responses by Targeting ADAM10 in Human THP-1 Monocytes. Mediat. Inflamm. 2019, 2019, 5306541. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.G.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig. Dis. Sci. 2012, 57, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Ristori, M.V.; Quagliariello, A.; Reddel, S.; Ianiro, G.; Vicari, S.; Gasbarrini, A.; Putignani, L. Autism, Gastrointestinal Symptoms and Modulation of Gut Microbiota by Nutritional Interventions. Nutrients 2019, 11, 2812. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB 2015, 29, 1395–1403. [Google Scholar] [CrossRef]
- Gauthier, J.; Bonnel, A.; St-Onge, J.; Karemera, L.; Laurent, S.; Mottron, L.; Fombonne, E.; Joober, R.; Rouleau, G.A. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 132, 74–75. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Butler, M.G.; Takahashi, T.N.; Miles, J.H. Do known mutations in neuroligin genes (NLGN3 and NLGN4) cause autism? J. Autism Dev. Disord. 2004, 34, 735–736. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Földy, C.; Malenka, R.C.; Südhof, T.C. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013, 78, 498–509. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ADAM10 Substrates | Shedding Reduction in ADAM10–/– Neurons |
|---|---|
| Cx3cl1 | 91% |
| NLGN-1 | 83% |
| PCDH9 | 71% |
| NrCAM | 66% |
| NLGN-3 | 62% |
| APP | 20% |
| Protein Name | Gene Symbol | ADAM10 Shedding | ADAM17 Shedding | ASD Patients |
|---|---|---|---|---|
| Amyloid Precursor Protein | APP | ↑sAPPα [32] | ↑sAPPα [32] | ↑ sAPPα [28] |
| Neuroligin-1 | NLGN-1 | ↓ Synaptogenic activity [92] | Common variants [249,250] | |
| Neuroligin-3 | NLGN-3 | ↓ Synaptogenic activity [92] | R415C transition [251,252] Common variants [250] | |
| Neurexin-1 | NRXN-1 | ↓ Synaptogenic activity [93] | Loss-of-function variants [24,26,27] | |
| Neural glial-related Cell Adhesion Molecule | NrCAM | ↑ Axon targeting activity [35] | SNPs & Common variants [25] | |
| Protocadherin9 | PCDH9 | No data available | Copy Number Variants [24] | |
| Fractalkine | CX3CL1 | ↑ Synaptic pruning [35,121,122] | No data available | |
| Tumor Necrosis Factor-α | TNF-α | ↑ pro-inflammatory activity [131] | ↑ in blood and brain [134] | |
| Interleukin-6 Receptor | IL-6R | ↑ pro-inflammatory pathways [139] | ↑ pro-inflammatory pathways [139] | ↑ IL-6 in blood and brain [134] |
| Triggering Receptor Expressed in Myeloid cells 2 | TREM2 | ↓ TREM2 membrane receptor levels [143] | ↓ in post-mortem brain tissue age 5–23 [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Verhoeff, T.A.; Perez Pardo, P.; Garssen, J.; Kraneveld, A.D. The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17. Int. J. Mol. Sci. 2021, 22, 118. https://doi.org/10.3390/ijms22010118
Zheng Y, Verhoeff TA, Perez Pardo P, Garssen J, Kraneveld AD. The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17. International Journal of Molecular Sciences. 2021; 22(1):118. https://doi.org/10.3390/ijms22010118
Chicago/Turabian StyleZheng, Yuanpeng, Tessa A. Verhoeff, Paula Perez Pardo, Johan Garssen, and Aletta D. Kraneveld. 2021. "The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17" International Journal of Molecular Sciences 22, no. 1: 118. https://doi.org/10.3390/ijms22010118
APA StyleZheng, Y., Verhoeff, T. A., Perez Pardo, P., Garssen, J., & Kraneveld, A. D. (2021). The Gut-Brain Axis in Autism Spectrum Disorder: A Focus on the Metalloproteases ADAM10 and ADAM17. International Journal of Molecular Sciences, 22(1), 118. https://doi.org/10.3390/ijms22010118