Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guidance and Neurological Disorders


Abstract
1. Axon Guidance
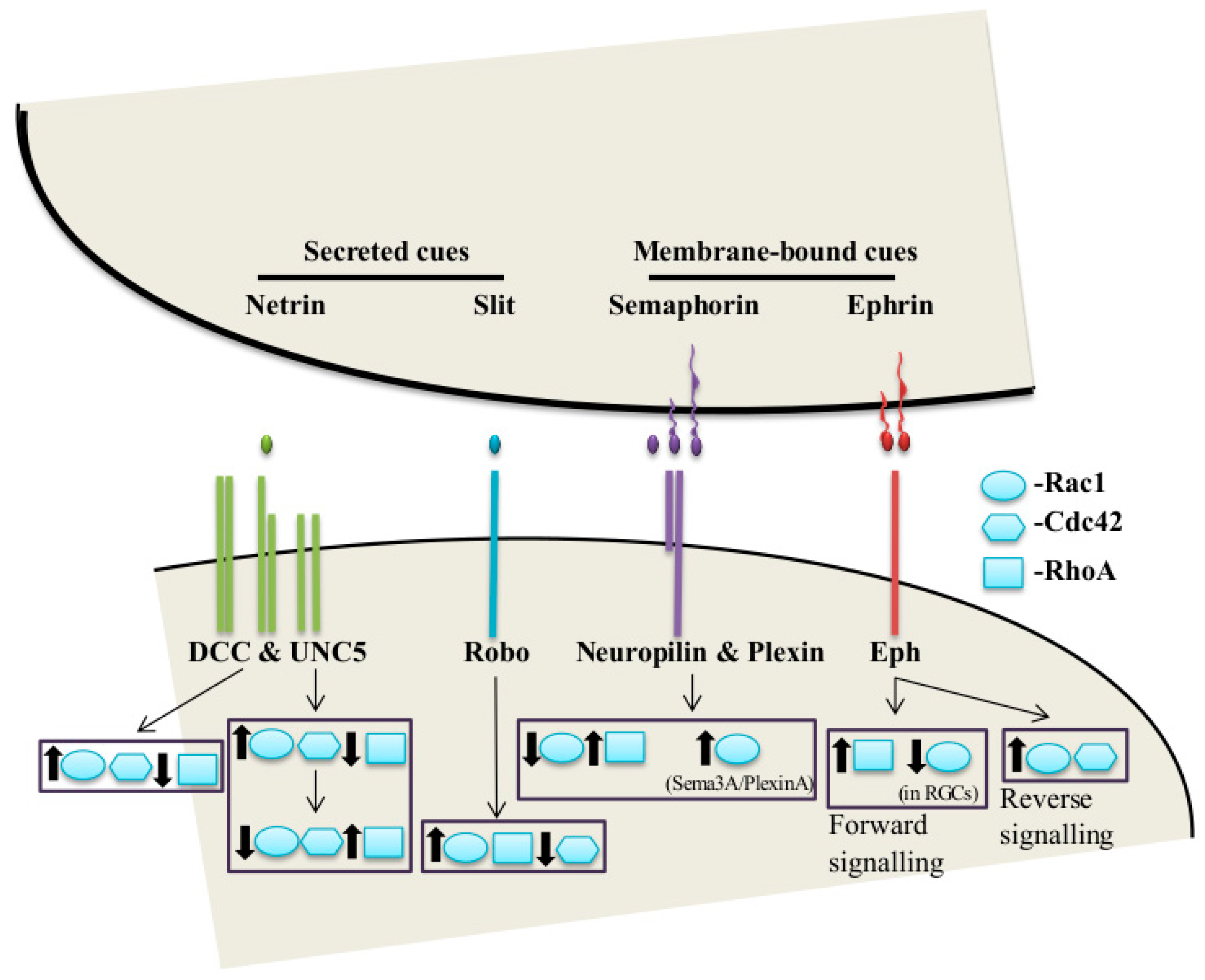
2. The Classical Guidance Cues and Their Receptors in Axon Guidance
2.1. Netrins
2.2. Slits
2.3. Semaphorins
2.4. Ephrins
3. Molecular Mechanisms of Axon Guidance
3.1. Regulation of Receptor Complexes
3.2. Trafficking and Surface Enrichment of Guidance Receptors
3.3. Regulated Endocytosis of Guidance Receptors
3.4. Proteolytic Processing of Guidance Receptors
3.5. Downstream Signaling from Axon Guidance Receptor Complexes
3.5.1. Calcium and Cyclic Nucleotides
3.5.2. Linking Axon Guidance Receptor Signaling to the Actin Cytoskeleton: Rho GTPases and Their Regulators—GAPs and GEFs
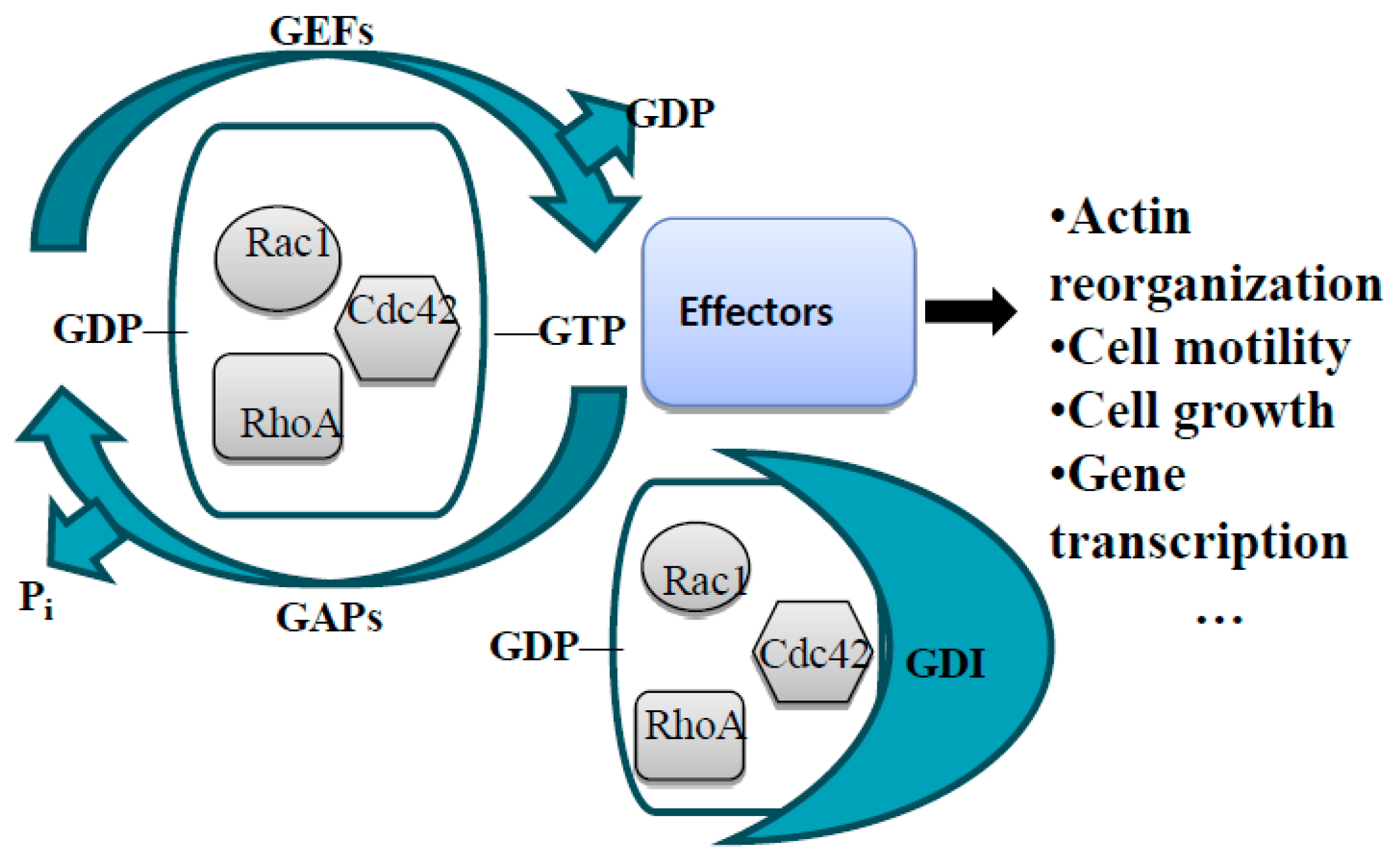
The Rho Family of Small GTPases in Guidance Receptor Signaling
General Information about GEFs and GAPs
RhoGAPs and GEFs in Axon Guidance
3.6. Netrin-1/DCC
3.7. Slit-Robo
3.8. Sema-Plexin
3.9. Ephrin-Eph
4. Regulators of Rho GTPases in Neurological Disorders
4.1. RhoGAPs
4.1.1. ARHGAP2 (α-Chimaerin/CHN1)
4.1.2. ARHGAP15
4.1.3. ARHGAP18 (MacGAP, SENEX)
4.1.4. ARHGAP28
4.1.5. ARHGAP32 (p250GAP)
4.1.6. ARHGAP14 (srGAP3) and ARHGAP34 (srGAP2)
4.1.7. ARHGAP33 (TCGAP)
4.1.8. ARHGAP41 (Oligophrenin-1/OPHN1)
4.1.9. ARHGAP43 (SH3BP1/3BP-1)
4.1.10. ARHGAP44 (RICH2/Nadrin1)
4.1.11. ARAP1, ARAP3, ARHGAP12, ARHGAP29, ARHGAP40, and ARHGAP45 (HMHA1)
4.1.12. ARHGAP46 (GMIP)
4.1.13. Myosin IXb (MYO9B)
4.1.14. Ral Binding Protein 1 (RalBP1/RLIP76)
4.2. RhoGEFs
4.2.1. ARHGEF2 (GEF-H1)
4.2.2. ARHGEF6 (αPIX/Cool-2)
4.2.3. ARHGEF9 (Collybistin)
4.2.4. ARHGEF10
4.2.5. ARHGEF13 (A-Kinase Anchor Protein 13 (AKAP13)/LBC/BRX)
4.2.6. ARHGEF23 (Trio)
4.2.7. ARHGEF24 (Kalirin)
4.2.8. ARHGEF36 (DNMBP)
4.2.9. ARHGEF42 (PLEKHG2)
4.2.10. ARHGEF44 (Puratrophin-1/PLEKHG4)
4.2.11. Alsin
4.2.12. Faciogenital Dysplasia Protein 1 (FGD1)
4.2.13. VAV1 and VAV3
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinez, E.; Tran, T.S. Vertebrate spinal commissural neurons: A model system for studying axon guidance beyond the midline. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 283–297. [Google Scholar] [CrossRef]
- Mueller, B.K. Growth cone guidance: First steps towards a deeper understanding. Annu. Rev. Neurosci. 1999, 22, 351–388. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.C.; Wadsworth, W.G. Axon guidance: Asymmetric signaling orients polarized outgrowth. Trends Cell Biol. 2008, 18, 597–603. [Google Scholar] [CrossRef]
- Zou, Y.; Lyuksyutova, A.I. Morphogens as conserved axon guidance cues. Curr. Opin. Neurobiol. 2007, 17, 22–28. [Google Scholar] [CrossRef]
- Chilton, J.K. Molecular mechanisms of axon guidance. Dev. Biol. 2006, 292, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Charron, F. Midline axon guidance and human genetic disorders. Clin. Genet. 2011, 80, 226–234. [Google Scholar] [CrossRef]
- Kaprielian, Z.; Runko, E.; Imondi, R. Axon guidance at the midline choice point. Dev. Dyn. 2001, 221, 154–181. [Google Scholar] [CrossRef]
- Chédotal, A.; Richards, L.J. Wiring the brain: The biology of neuronal guidance. Cold Spring Harb. Perspect. Biol. 2010, 2, a001917. [Google Scholar] [CrossRef]
- Y Cajal, S.R. Histology of the Nervous System of Man and Vertebrates; Oxford University Press: Oxford, UK, 1995; Volume 1. [Google Scholar]
- Ramon, Y.; Cajal, S. Textura del Sistema Nervioso del Hombre y de los Vertebrados; Madrid Nicolas Moya: Madrid, Spain, 1904; Volume 2. [Google Scholar]
- Raper, J.; Mason, C. Cellular strategies of axonal pathfinding. Cold Spring Harb. Perspect. Biol. 2010, 2, a001933. [Google Scholar] [CrossRef]
- Harrison, R.G. Observations on the living developing nerve fiber. Proc. Soc. Exp. Biol. Med. 1907, 4, 140–143. [Google Scholar] [CrossRef]
- Harrison, R.G. Further experiments on the development of peripheral nerves. Dev. Dyn. 1906, 5, 121–131. [Google Scholar] [CrossRef]
- Harrison, R.G. The outgrowth of the nerve fiber as a mode of protoplasmic movement. J. Exp. Zool. Part A Ecol. Genet. Physiol. 1910, 9, 787–846. [Google Scholar] [CrossRef]
- Sperry, R.W. Chemoaffinity in the orderly growth of nerve fiber patterns and connections. Proc. Natl. Acad. Sci. USA 1963, 50, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin, A.L.; Tessier-Lavigne, M. Mechanisms and molecules of neuronal wiring: A primer. Cold Spring Harb. Perspect. Biol. 2011, 3, a001727. [Google Scholar] [CrossRef] [PubMed]
- Yaron, A.; Zheng, B. Navigating their way to the clinic: Emerging roles for axon guidance molecules in neurological disorders and injury. Dev. Neurobiol. 2007, 67, 1216–1231. [Google Scholar] [CrossRef]
- Huber, A.B.; Kolodkin, A.L.; Ginty, D.D.; Cloutier, J.-F. Signaling at the growth cone: Ligand-receptor complexes and the control of axon growth and guidance. Annu. Rev. Neurosci. 2003, 26, 509–563. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, M.A.; Cowan, C.W. Signaling mechanisms of axon guidance and early synaptogenesis. In The Neurobiology of Childhood; Springer: Berlin, Germany, 2013; pp. 19–48. [Google Scholar]
- Van Battum, E.Y.; Brignani, S.; Pasterkamp, R.J. Axon guidance proteins in neurological disorders. Lancet Neurol. 2015, 14, 532–546. [Google Scholar] [CrossRef]
- Lin, L.; Lesnick, T.G.; Maraganore, D.M.; Isacson, O. Axon guidance and synaptic maintenance: Preclinical markers for neurodegenerative disease and therapeutics. Trends Neurosci. 2009, 32, 142–149. [Google Scholar] [CrossRef]
- Adams, R.H.; Eichmann, A. Axon guidance molecules in vascular patterning. Cold Spring Harb. Perspect. Biol. 2010, 2, a001875. [Google Scholar] [CrossRef]
- Cirulli, V.; Yebra, M. Netrins: Beyond the brain. Nat. Rev. Mol. Cell Biol. 2007, 8, 296–306. [Google Scholar] [CrossRef]
- Moore, S.W.; Tessier-Lavigne, M.; Kennedy, T.E. Netrins and their receptors. In Axon Growth and Guidance; Springer: Berlin, Germany, 2007; pp. 17–31. [Google Scholar]
- Yamagishi, S.; Yamada, K.; Sawada, M.; Nakano, S.; Mori, N.; Sawamoto, K.; Sato, K. Netrin-5 is highly expressed in neurogenic regions of the adult brain. Front. Cell Neurosci. 2015, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Dominici, C.; Moreno-Bravo, J.A.; Puiggros, S.R.; Rappeneau1, Q.; Rama, N.; Vieugue, P.; Bernet, A.; Mehlen, P.; Chédotal, A. Floor plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 2017, 545, 350–354. [Google Scholar] [CrossRef]
- Yamauchi, K.; Yamazaki, M.; Abe, M.; Sakimura, K.; Lickert, H.; Kawasaki, T.; Murakami, F.; Hirata, T. Netrin-1 derived from the ventricular zone, but not the floor plate, directs hindbrain commissural axons to the ventral midline. Sci. Rep. 2017, 7, 1192. [Google Scholar] [CrossRef]
- Varadarajan, S.G.; Kong, J.H.; Phan, K.D.; Kao, T.J.; Panaitof, S.C.; Cardin, J.; Eltzschig, H.; Kania, A.; Novitch, B.G.; Butler, S.J. Netrin-1 produced by neural progenitors, not floor plate cells, is required for axon guidance in the spinal cord. Neuron 2017, 94, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bravo, J.A.; SPuiggros, R.; Mehlen, P.; Chedotal, A. Synergistic activity of floor plate and ventricular zone-derived netrin-1 in spinal cord commissural axon guidance. Neuron 2019, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Hinck, L.; Nishiyama, M.; Poo, M.-M.; Tessier-Lavigne, M.; Stein, E. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 1999, 97, 927–941. [Google Scholar] [CrossRef]
- Merz, D.C.; Zheng, H.; Killeen, M.T.; Krizus, A.; Culotti, J.G. Multiple signaling mechanisms of the UNC-6/netrin receptors UNC-5 and UNC-40/DCC in vivo. Genetics 2001, 158, 1071–1080. [Google Scholar]
- Walter, J.; Henke-Fahle, S.; Bonhoeffer, F. Avoidance of posterior tectal membranes by temporal retinal axons. Development 1987, 101, 909–913. [Google Scholar]
- Drescher, U.; Kremoser, C.; Handwerker, C.; Löschinger, J.; Noda, M.; Bonhoeffer, F. In vitro guidance of retinal ganglion cell axons by RAGS, a 25 kDa tectal protein related to ligands for Eph receptor tyrosine kinases. Cell 1995, 82, 359–370. [Google Scholar] [CrossRef]
- Antoine-Bertrand, J.; Villemure, J.-F.; Lamarche-Vane, N. Implication of Rho GTPases in neurodegenerative diseases. Curr. Drug Targets 2011, 12, 1202–1215. [Google Scholar] [CrossRef]
- DeGeer, J.; Kaplan, A.; Mattar, P.; Morabito, M.; Stochaj, U.; Kennedy, T.E.; Debant, A.; Cayouette, M.; Fournier, A.E.; Lamarche-Vane, N. Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J. Cell Biol. 2015, 210, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Bashaw, G.J.; Klein, R. Signaling from axon guidance receptors. Cold Spring Harb. Perspect. Biol. 2010, 2, a001941. [Google Scholar] [CrossRef] [PubMed]
- Gomez, T.M.; Zheng, J.Q. The molecular basis for calcium-dependent axon pathfinding. Nat. Rev. Neurosci. 2006, 7, 115. [Google Scholar] [CrossRef]
- Zheng, J.Q.; Poo, M.-M. Calcium signaling in neuronal motility. Annu. Rev. Cell Dev. Biol. 2007, 23, 375–404. [Google Scholar] [CrossRef]
- Henley, J.; Poo, M.-M. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, Y.-C.; Cui, K.; Li, N.; Zheng, Z.-Y.; Wang, Y.-Z.; Yuan, X.-B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 2005, 434, 894. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; von Schimmelmann, M.J.; Togashi, K.; Findley, W.M.; Hong, K. Membrane potential shifts caused by diffusible guidance signals direct growth-cone turning. Nat. Neurosci. 2008, 11, 762. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.X.; Poo, M.-M. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature 2005, 434, 898. [Google Scholar] [CrossRef]
- Nishiyama, M.; Hoshino, A.; Tsai, L.; Henley, J.R.; Goshima, Y.; Tessier-Lavigne, M.; Poo, M.-M.; Hong, K. Cyclic AMP/GMP-dependent modulation of Ca 2+ channels sets the polarity of nerve growth-cone turning. Nature 2003, 423, 990. [Google Scholar] [CrossRef]
- Song, H.-J.; Ming, G.-L.; He, Z.; Lehmann, M.; McKerracher, L.; Tessier-Lavigne, M.; Poo, M.-M. Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 1998, 281, 1515–1518. [Google Scholar] [CrossRef]
- Song, H.-J.; Ming, G.-L.; Poo, M.-M. cAMP-induced switching in turning direction of nerve growth cones. Nature 1997, 388, 275. [Google Scholar] [CrossRef]
- Tojima, T.; Hines, J.H.; Henley, J.R.; Kamiguchi, H. Second messengers and membrane trafficking direct and organize growth cone steering. Nat. Rev. Neurosci. 2011, 12, 191–203. [Google Scholar] [CrossRef]
- Dent, E.W.; Tang, F.; Kalil, K. Axon guidance by growth cones and branches: Common cytoskeletal and signaling mechanisms. Neuroscientist 2003, 9, 343–353. [Google Scholar] [CrossRef]
- Dickson, B.J. Molecular mechanisms of axon guidance. Science 2002, 298, 1959–1964. [Google Scholar] [CrossRef]
- Hall, A.; Lalli, G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2010, 2, a001818. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.W.; Flynn, K.C.; Bamburg, J.R. Actin-binding proteins take the reins in growth cones. Nat. Rev. Neurosci. 2008, 9, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Boyer, N.P.; Gupton, S.L. Revisiting netrin-1: One who guides (axons). Front. Cell. Neurosci. 2018, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef]
- Mackay, D.J.; Esch, F.; Furthmayr, H.; Hall, A. Rho-and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: An essential role for ezrin/radixin/moesin proteins. J. Cell Biol. 1997, 138, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999, 144, 1235–1244. [Google Scholar] [CrossRef]
- Li, X.; Saint-Cyr-Proulx, E.; Aktories, K.; Lamarche-Vane, N. Rac1 and Cdc42 but not RhoA or Rho kinase activities are required for neurite outgrowth induced by the Netrin-1 receptor DCC (deleted in colorectal cancer) in N1E-115 neuroblastoma cells. J. Biol. Chem. 2002, 277, 15207–15214. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.W.; Correia, J.P.; Sun, K.L.W.; Pool, M.; Fournier, A.E.; Kennedy, T.E. Rho inhibition recruits DCC to the neuronal plasma membrane and enhances axon chemoattraction to netrin 1. Development 2008, 135, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- Shekarabi, M.; Kennedy, T.E. The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol. Cell. Neurosci. 2002, 19, 1–17. [Google Scholar] [CrossRef]
- Keleman, K.; Dickson, B.J. Short-and long-range repulsion by the Drosophila Unc5 netrin receptor. Neuron 2001, 32, 605–617. [Google Scholar] [CrossRef]
- Picard, M.; Petrie, R.J.; Antoine-Bertrand, J.; Saint-Cyr-Proulx, E.; Villemure, J.-F.; Lamarche-Vane, N. Spatial and temporal activation of the small GTPases RhoA and Rac1 by the netrin-1 receptor UNC5a during neurite outgrowth. Cell. Signal. 2009, 21, 1961–1973. [Google Scholar] [CrossRef]
- Hu, H.; Marton, T.F.; Goodman, C.S. Plexin B mediates axon guidance in Drosophila by simultaneously inhibiting active Rac and enhancing RhoA signaling. Neuron 2001, 32, 39–51. [Google Scholar] [CrossRef]
- Swiercz, J.M.; Kuner, R.; Behrens, J.; Offermanns, S. Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 2002, 35, 51–63. [Google Scholar] [CrossRef]
- Turner, L.J.; Nicholls, S.; Hall, A. The activity of the plexin-A1 receptor is regulated by Rac. J. Biol. Chem. 2004, 279, 33199–33205. [Google Scholar] [CrossRef]
- Fan, X.; Labrador, J.P.; Hing, H.; Bashaw, G.J. Slit stimulation recruits Dock and Pak to the roundabout receptor and increases Rac activity to regulate axon repulsion at the CNS midline. Neuron 2003, 40, 113–127. [Google Scholar] [CrossRef]
- Wong, K.; Ren, X.-R.; Huang, Y.-Z.; Xie, Y.; Liu, G.; Saito, H.; Tang, H.; Wen, L.; Brady-Kalnay, S.M.; Mei, L. Signal transduction in neuronal migration: Roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell 2001, 107, 209–221. [Google Scholar] [CrossRef]
- Jurney, W.M.; Gallo, G.; Letourneau, P.C.; McLoon, S.C. Rac1-mediated endocytosis during ephrin-A2-and semaphorin 3A-induced growth cone collapse. J. Neurosci. 2002, 22, 6019–6028. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Barth, H.; Ciossek, T.; Aktories, K.; Mueller, B.K. Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J. Cell Biol. 2000, 149, 263–270. [Google Scholar] [CrossRef]
- Xu, N.-J.; Henkemeyer, M. Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat. Neurosci. 2009, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.D.; Self, A.J.; van Oers, C.; Hall, A. Identification of distinct cytoplasmic targets for ras/R-ras and rho regulatory proteins. J. Biol. Chem. 1989, 264, 10–13. [Google Scholar] [PubMed]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Xiang, X.; Liang, C.; Shi, L. Regulating Rac in the nervous system: Molecular function and disease implication of Rac GEFs and GAPs. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A. Coevolution of RAC Small GTPases and their Regulators GEF Proteins. Evol. Bioinform. Online 2016, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Tcherkezian, J.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Laurin, M.; Côté, J.-F. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014, 28, 533–547. [Google Scholar] [CrossRef]
- Namekata, K.; Kimura, A.; Kawamura, K.; Harada, C.; Harada, T. Dock GEFs and their therapeutic potential: Neuroprotection and axon regeneration. Prog. Retin. Eye Res. 2014, 43, 1–16. [Google Scholar] [CrossRef]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Shi, L. Dock protein family in brain development and neurological disease. Commun. Integr. Biol. 2013, 6, e26839. [Google Scholar] [CrossRef] [PubMed]
- Peck, J.; Douglas, G.; Wu, C.H.; Burbelo, P.D. Human RhoGAP domain-containing proteins: Structure, function and evolutionary relationships. Febs Lett. 2002, 528, 27–34. [Google Scholar] [CrossRef]
- DeGeer, J.; Boudeau, J.; Schmidt, S.; Bedford, F.; Lamarche-Vane, N.; Debant, A. Tyrosine phosphorylation of the RhoGEF Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol. Cell. Biol. 2013, 33, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Tata, A.; Stoppel, D.C.; Hong, S.; Ben-Zvi, A.; Xie, T.; Gu, C. An image-based RNAi screen identifies SH3BP1 as a key effector of Semaphorin 3E–PlexinD1 signaling. J. Cell Biol. 2014, 205, 573–590. [Google Scholar] [CrossRef]
- Brouns, M.R.; Matheson, S.F.; Settleman, J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat. Cell Biol. 2001, 3, 361–368. [Google Scholar] [CrossRef]
- Briançon-Marjollet, A.; Ghogha, A.; Nawabi, H.; Triki, I.; Auziol, C.; Fromont, S.; Piché, C.; Enslen, H.; Chebli, K.; Cloutier, J.-F. Trio mediates netrin-1-induced Rac1 activation in axon outgrowth and guidance. Mol. Cell. Biol. 2008, 28, 2314–2323. [Google Scholar] [CrossRef]
- Forsthoefel, D.J.; Liebl, E.C.; Kolodziej, P.A.; Seeger, M.A. The Abelson tyrosine kinase, the Trio GEF and Enabled interact with the Netrin receptor Frazzled in Drosophila. Development 2005, 132, 1983–1994. [Google Scholar] [CrossRef]
- Li, X.; Gao, X.; Liu, G.; Xiong, W.; Wu, J.; Rao, Y. Netrin signal transduction and the guanine nucleotide exchange factor DOCK180 in attractive signaling. Nat. Neurosci. 2008, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sommer, J.E.; Martin, J.H.; Scheiffele, P. α2-Chimaerin is an essential EphA4 effector in the assembly of neuronal locomotor circuits. Neuron 2007, 55, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Iwasato, T.; Katoh, H.; Nishimaru, H.; Ishikawa, Y.; Inoue, H.; Saito, Y.M.; Ando, R.; Iwama, M.; Takahashi, R.; Negishi, M. Rac-GAP α-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell 2007, 130, 742–753. [Google Scholar] [CrossRef]
- Kullander, K.; Croll, S.D.; Zimmer, M.; Pan, L.; McClain, J.; Hughes, V.; Zabski, S.; DeChiara, T.M.; Klein, R.; Yancopoulos, G.D. Ephrin-B3 is the midline barrier that prevents corticospinal tract axons from recrossing, allowing for unilateral motor control. Genes Dev. 2001, 15, 877–888. [Google Scholar] [CrossRef]
- Shi, L.; Fu, W.-Y.; Hung, K.-W.; Porchetta, C.; Hall, C.; Fu, A.K.; Ip, N.Y. α2-chimaerin interacts with EphA4 and regulates EphA4-dependent growth cone collapse. Proc. Natl. Acad. Sci. USA 2007, 104, 16347–16352. [Google Scholar] [CrossRef]
- Wegmeyer, H.; Egea, J.; Rabe, N.; Gezelius, H.; Filosa, A.; Enjin, A.; Varoqueaux, F.; Deininger, K.; Schnütgen, F.; Brose, N. EphA4-dependent axon guidance is mediated by the RacGAP α2-chimaerin. Neuron 2007, 55, 756–767. [Google Scholar] [CrossRef]
- Cowan, C.W.; Shao, Y.R.; Sahin, M.; Shamah, S.M.; Lin, M.Z.; Greer, P.L.; Gao, S.; Griffith, E.C.; Brugge, J.S.; Greenberg, M.E. Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron 2005, 46, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Shamah, S.M.; Lin, M.Z.; Goldberg, J.L.; Estrach, S.; Sahin, M.; Hu, L.; Bazalakova, M.; Neve, R.L.; Corfas, G.; Debant, A. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 2001, 105, 233–244. [Google Scholar] [CrossRef]
- Toyofuku, T.; Yoshida, J.; Sugimoto, T.; Zhang, H.; Kumanogoh, A.; Hori, M.; Kikutani, H. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat. Neurosci. 2005, 8, 1712. [Google Scholar] [CrossRef]
- Barberis, D.; Casazza, A.; Sordella, R.; Corso, S.; Artigiani, S.; Settleman, J.; Comoglio, P.M.; Tamagnone, L. p190 Rho-GTPase activating protein associates with plexins and it is required for semaphorin signalling. J. Cell Sci. 2005, 118, 4689–4700. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, M.; Labrador, J.-P.; McEwen, J.; Lai, E.C.; Goodman, C.S.; Bashaw, G.J. Cross GTPase-activating protein (CrossGAP)/Vilse links the Roundabout receptor to Rac to regulate midline repulsion. Proc. Natl. Acad. Sci. USA 2005, 102, 4613–4618. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.L.; van Berkum, M. Calmodulin and son of sevenless dependent signaling pathways regulate midline crossing of axons in the Drosophila CNS. Development 2000, 127, 1991–2000. [Google Scholar] [PubMed]
- Yang, L.; Bashaw, G.J. Son of sevenless directly links the Robo receptor to rac activation to control axon repulsion at the midline. Neuron 2006, 52, 595–607. [Google Scholar] [CrossRef]
- Wu, W.; Wong, K.; Chen, J.-H.; Jiang, Z.-H.; Dupuis, S.; Wu, J.Y.; Rao, Y. Directional guidance of neuronal migration in the olfactory system by the protein Slit. Nature 1999, 400, 331. [Google Scholar] [CrossRef]
- Debant, A.; Serra-Pages, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate Rac-specific and Rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef]
- Lundström, A.; Gallio, M.; Englund, C.; Steneberg, P.; Hemphälä, J.; Aspenström, P.; Keleman, K.; Falileeva, L.; Dickson, B.J.; Samakovlis, C. Vilse, a conserved Rac/Cdc42 GAP mediating Robo repulsion in tracheal cells and axons. Genes Dev. 2004, 18, 2161–2171. [Google Scholar] [CrossRef]
- Vikis, H.G.; Li, W.; Guan, K.-L. The plexin-B1/Rac interaction inhibits PAK activation and enhances Sema4D ligand binding. Genes Dev. 2002, 16, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Strittmatter, S.M. Rac1 mediates collapsin-1-induced growth cone collapse. J. Neurosci. 1997, 17, 6256–6263. [Google Scholar] [CrossRef] [PubMed]
- Kullander, K.; Butt, S.J.; Lebret, J.M.; Lundfald, L.; Restrepo, C.E.; Rydström, A.; Klein, R.; Kiehn, O. Role of EphA4 and EphrinB3 in local neuronal circuits that control walking. Science 2003, 299, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.B.; Brown, M.D.; Wilcox, C.L.; Raper, J.A.; Bamburg, J.R. Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: Inhibition of collapse by opposing mutants of rac1. J. Neurosci. 1999, 19, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Västrik, I.; Eickholt, B.J.; Walsh, F.S.; Ridley, A.; Doherty, P. Sema3A-induced growth-cone collapse is mediated by Rac1 amino acids 17–32. Curr. Biol. 1999, 9, 991–998. [Google Scholar] [CrossRef]
- Marston, D.J.; Dickinson, S.; Nobes, C.D. Rac-dependent trans-endocytosis of ephrinBs regulates Eph–ephrin contact repulsion. Nat. Cell Biol. 2003, 5, 879. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Palmer, A.; Köhler, J.; Klein, R. EphB–ephrinB bi-directional endocytosis terminates adhesion allowing contact mediated repulsion. Nat. Cell Biol. 2003, 5, 869. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Orioli, D.; Henderson, J.T.; Saxton, T.M.; Roder, J.; Pawson, T.; Klein, R. Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell 1996, 86, 35–46. [Google Scholar] [CrossRef]
- Logotheti, M.; Papadodima, O.; Chatziioannou, A.; Venizelos, N.; Kolisis, F. Gene Expression Analysis of Fibroblasts from Patients with Bipolar Disorder. J. Neuropsychopharmacol. Ment. Health 2015, 1, 1–9. [Google Scholar]
- Iwata, R.; Ohi, K.; Kobayashi, Y.; Masuda, A.; Iwama, M.; Yasuda, Y.; Yamamori, H.; Tanaka, M.; Hashimoto, R.; Itohara, S. RacGAP α2-Chimaerin Function in Development Adjusts Cognitive Ability in Adulthood. Cell Rep. 2014, 8, 1257–1264. [Google Scholar] [CrossRef]
- Zamboni, V.; Armentano, M.; Sarò, G.; Ciraolo, E.; Ghigo, A.; Germena, G.; Umbach, A.; Valnegri, P.; Passafaro, M.; Carabelli, V. Disruption of ArhGAP15 results in hyperactive Rac1, affects the architecture and function of hippocampal inhibitory neurons and causes cognitive deficits. Sci. Rep. 2016, 6, 34877. [Google Scholar] [CrossRef]
- Peng, Y.-J.; He, W.-Q.; Tang, J.; Tao, T.; Chen, C.; Gao, Y.-Q.; Zhang, W.-C.; He, X.-Y.; Dai, Y.-Y.; Zhu, N.-C. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem. 2010, 285, 24834–24844. [Google Scholar] [CrossRef]
- Cahill, M.E.; Xie, Z.; Day, M.; Photowala, H.; Barbolina, M.V.; Miller, C.A.; Weiss, C.; Radulovic, J.; Sweatt, J.D.; Disterhoft, J.F. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc. Natl. Acad. Sci. USA 2009, 106, 13058–13063. [Google Scholar] [CrossRef]
- Hill, J.; Hashimoto, T.; Lewis, D. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Krug, T.; Manso, H.; Gouveia, L.; Sobral, J.; Xavier, J.M.; Albergaria, I.; Gaspar, G.; Correia, M.; Viana-Baptista, M.; Simões, R.M. Kalirin: A novel genetic risk factor for ischemic stroke. Hum. Genet. 2010, 127, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Mandela, P.; Ma, X.-M. Kalirin, a key player in synapse formation, is implicated in human diseases. Neural Plasticity 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Macciardi, F.; Guffanti, G.; Fallon, J.H.; Wang, Q.; Turner, J.A.; Lakatos, A.; Miles, M.F.; Lander, A.; Vawter, M.P. Identifying gene regulatory networks in schizophrenia. Neuroimage 2010, 53, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Turner, J.; Fallon, J.; Lakatos, A.; Keator, D.; Guffanti, G.; Macciardi, F. Gene discovery through imaging genetics: Identification of two novel genes associated with schizophrenia. Mol. Psychiatry 2009, 14, 416–428. [Google Scholar] [CrossRef]
- Guo, W.; Cai, Y.; Zhang, H.; Yang, Y.; Yang, G.; Wang, X.; Zhao, J.; Lin, J.; Zhu, J.; Li, W. Association of ARHGAP18 polymorphisms with schizophrenia in the Chinese-Han population. PLoS ONE 2017, 12, e0175209. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Rivalan, M.; Strauss, U.; Stoenica, L.; Trimbuch, T.; Rademacher, N.; Parthasarathy, S.; Lajko, D.; Rosenmund, C.; Shoichet, S. NOMA-GAP/ARHGAP33 regulates synapse development and autistic-like behavior in the mouse. Mol. Psychiatry 2015, 20, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Hashimoto, R.; Sakoori, K.; Sugaya, Y.; Tanimura, A.; Hashimotodani, Y.; Ohi, K.; Yamamori, H.; Yasuda, Y.; Umeda-Yano, S. Emerging roles of ARHGAP33 in intracellular trafficking of TrkB and pathophysiology of neuropsychiatric disorders. Nat. Commun. 2016, 7, 10594. [Google Scholar] [CrossRef] [PubMed]
- Jungerius, B.J.; Bakker, S.C.; Monsuur, A.J.; Sinke, R.J.; Kahn, R.S.; Wijmenga, C. Is MYO9B the missing link between schizophrenia and celiac disease? Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 351–355. [Google Scholar] [CrossRef]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F. Increased burden of ultra-rare protein-altering variants among 4877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433. [Google Scholar] [CrossRef]
- Ikeda, M.; Aleksic, B.; Kinoshita, Y.; Okochi, T.; Kawashima, K.; Kushima, I.; Ito, Y.; Nakamura, Y.; Kishi, T.; Okumura, T. Genome-wide association study of schizophrenia in a Japanese population. Biol. Psychiatry 2011, 69, 472–478. [Google Scholar] [CrossRef]
- Aleksic, B.; Kushima, I.; Hashimoto, R.; Ohi, K.; Ikeda, M.; Yoshimi, A.; Nakamura, Y.; Ito, Y.; Okochi, T.; Fukuo, Y. Analysis of the VAV3 as candidate gene for schizophrenia: Evidences from voxel-based morphometry and mutation screening. Schizophr. Bull. 2012, 39, 720–728. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, R.; Chen, C.; You, Z.-F.; Luo, X.; Wang, X.-P. Six novel rare non-synonymous mutations for migraine without aura identified by exome sequencing. J. Neurogenet. 2015, 29, 188–194. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef]
- Varma, H.; Yamamoto, A.; Sarantos, M.R.; Hughes, R.E.; Stockwell, B.R. Mutant huntingtin alters cell fate in response to microtubule depolymerization via the GEF-H1-RhoA-ERK pathway. J. Biol. Chem. 2010, 285, 37445–37457. [Google Scholar] [CrossRef]
- Sarowar, T.; Grabrucker, S.; Föhr, K.; Mangus, K.; Eckert, M.; Bockmann, J.; Boeckers, T.M.; Grabrucker, A.M. Enlarged dendritic spines and pronounced neophobia in mice lacking the PSD protein RICH2. Mol. Brain 2016, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Bhat, G.; LaGrave, D.; Millson, A.; Herriges, J.; Lamb, A.N.; Matalon, R. Xq11. 1-11.2 deletion involving ARHGEF9 in a girl with autism spectrum disorder. Eur. J. Med. Genet. 2016, 59, 470–473. [Google Scholar] [CrossRef]
- Poelmans, G.; Franke, B.; Pauls, D.; Glennon, J.; Buitelaar, J. AKAPs integrate genetic findings for autism spectrum disorders. Transl. Psychiatry 2013, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Sciience 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237. [Google Scholar] [CrossRef]
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonen, I.J.; Brouwer, A.P.; Brunner, H.G.; Webber, C. Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 2013, 34, 1679–1687. [Google Scholar] [CrossRef]
- Tadokoro, K.; Hashimoto, R.; Tatsumi, M.; Kosuga, A.; Kamijima, K.; Kunugi, H. The Gem interacting protein (GMIP) gene is associated with major depressive disorder. Neurogenetics 2005, 6, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.R.; Lloyd, K.E.; Kruszewski, A.; Kim, I.-H.; Rodriguiz, R.M.; Heindel, C.; Faytell, M.; Dudek, S.M.; Wetsel, W.C.; Soderling, S.H. WRP/srGAP3 facilitates the initiation of spine development by an inverse F-BAR domain, and its loss impairs long-term memory. J. Neurosci. 2011, 31, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Endris, V.; Wogatzky, B.; Leimer, U.; Bartsch, D.; Zatyka, M.; Latif, F.; Maher, E.R.; Tariverdian, G.; Kirsch, S.; Karch, D. The novel Rho-GTPase activating gene MEGAP/srGAP3 has a putative role in severe mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 11754–11759. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Zerres, K.; Senderek, J.; Rudnik-Schöneborn, S.; Eggermann, T.; Häusler, M.; Mull, M.; Ramaekers, V.T. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 2003, 126, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Billuart, P.; Bienvenu, T.; Ronce, N.; Portes, V.D.; Vinet, M.C.; Zemni, R.; Crollius, H.R.; Carrié, A.; Fauchereau, F.; Cherry, M. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 1998, 392, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rebouças, C.B.; Belet, S.; de Almeida, L.G.; Ribeiro, M.G.; Medina-Acosta, E.; Bahia, P.R.V.; da Silva, A.F.A.; Santos, F.L.D.; de Lacerda, G.C.B.; Pimentel, M.M.G. A novel in-frame deletion affecting the BAR domain of OPHN1 in a family with intellectual disability and hippocampal alterations. Eur. J. Hum. Genet. 2014, 22, 644–651. [Google Scholar] [CrossRef]
- Bedeschi, M.F.; Novelli, A.; Bernardini, L.; Parazzini, C.; Bianchi, V.; Torres, B.; Natacci, F.; Giuffrida, M.G.; Ficarazzi, P.; Dallapiccola, B. Association of syndromic mental retardation with an Xq12q13. 1 duplication encompassing the oligophrenin 1 gene. Am. J. Med. Genet. Part A 2008, 146, 1718–1724. [Google Scholar] [CrossRef]
- Philip, N.; Chabrol, B.; Lossi, A.; Cardoso, C.; Guerrini, R.; Dobyns, W.; Raybaud, C.; Villard, L. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J. Med. Genet. 2003, 40, 441–446. [Google Scholar] [CrossRef]
- Portes, V.D.; Boddaert, N.; Sacco, S.; Briault, S.; Maincent, K.; Bahi, N.; Gomot, M.; Ronce, N.; Bursztyn, J.; Adamsbaum, C. Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am. J. Med. Genet. Part A 2004, 124, 364–371. [Google Scholar] [CrossRef]
- Ravindran, E.; Hu, H.; Yuzwa, S.A.; Hernandez-Miranda, L.R.; Kraemer, N.; Ninnemann, O.; Musante, L.; Boltshauser, E.; Schindler, D.; Hübner, A. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. 2017, 13, e1006746. [Google Scholar] [CrossRef]
- Ramakers, G.J.; Wolfer, D.; Rosenberger, G.; Kuchenbecker, K.; Kreienkamp, H.-J.; Prange-Kiel, J.; Rune, G.; Richter, K.; Langnaese, K.; Masneuf, S. Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 2011, 21, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Kutsche, K.; Yntema, H.; Brandt, A.; Jantke, I.; Nothwang, H.G.; Orth, U.; Boavida, M.G.; David, D.; Chelly, J.; Fryns, J.-P. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 2000, 26, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Musante, L.; Fang, C.; Hoffmann, K.; Fuchs, C.; Carta, E.; Deas, E.; Venkateswarlu, K.; Menzel, C.; Ullmann, R. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 2009, 30, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.M.; Pendziwiat, M.; Eilam, A.; Gilad, R.; Blatt, I.; Rosenow, F.; Kanaan, M.; Helbig, I.; Afawi, Z. Israeli-Palestinian Epilepsy Family Consortium. The phenotypic spectrum of ARHGEF9 includes intellectual disability, focal epilepsy and febrile seizures. J. Neurol. 2017, 264, 1421–1425. [Google Scholar] [CrossRef]
- Long, P.; May, M.M.; James, V.M.; Grannò, S.; Johnson, J.P.; Tarpey, P.; Stevenson, R.E.; Harvey, K.; Schwartz, C.E.; Harvey, R.J. Missense mutation R338W in ARHGEF9 in a family with X-linked intellectual disability with variable macrocephaly and macro-orchidism. Front. Mol. Neurosci. 2016, 8, 83. [Google Scholar] [CrossRef]
- Marco, E.J.; Abidi, F.E.; Bristow, J.; Dean, W.B.; Cotter, P.; Jeremy, R.J.; Schwartz, C.E.; Sherr, E.H. ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J. Med. Genet. 2008, 45, 100–105. [Google Scholar] [CrossRef]
- Shimojima, K.; Sugawara, M.; Shichiji, M.; Mukaida, S.; Takayama, R.; Imai, K.; Yamamoto, T. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet. 2011, 56, 561–565. [Google Scholar] [CrossRef]
- Ba, W.; Yan, Y.; Reijnders, M.R.; Schuurs-Hoeijmakers, J.H.; Feenstra, I.; Bongers, E.M.; Bosch, D.G.; de Leeuw, N.; Pfundt, R.; Gilissen, C. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2015, 25, 892–902. [Google Scholar] [CrossRef]
- Katrancha, S.M.; Wu, Y.; Zhu, M.; Eipper, B.A.; Koleske, A.J.; Mains, R.E. Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum. Mol. Genet. 2017, 26, 4728–4740. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Pengelly, R.J.; Greville-Heygate, S.; Schmidt, S.; Seaby, E.G.; Jabalameli, M.R.; Mehta, S.G.; Parker, M.J.; Goudie, D.; Fagotto-Kaufmann, C.; Mercer, C.; et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 2016, 53, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Lebel, R.; May, M.; Pouls, S.; Lubs, H.; Stevenson, R.; Schwartz, C. Non-syndromic X-linked mental retardation associated with a missense mutation (P312L) in the FGD1 gene. Clin. Genet. 2002, 61, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.-S.; Chung, W.; Han, K.; Park, K.Y.; Kim, H.; Kim, E.; Kim, M.-H. Down-regulation of RalBP1 expression reduces seizure threshold and synaptic inhibition in mice. Biochem. Biophys. Res. Commun. 2013, 433, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.; Duguid, I.C.; Alldred, M.J.; Beatty, S.E.; Ward, H.; Keep, N.H.; Lingenfelter, S.E.; Pearce, B.R.; Lundgren, J.; Owen, M.J. The GDP-GTP exchange factor collybistin: An essential determinant of neuronal gephyrin clustering. J. Neurosci. 2004, 24, 5816–5826. [Google Scholar] [CrossRef]
- Kato, T.; Konishi, Y.; Shimohama, S.; Beach, T.G.; Akatsu, H.; Tooyama, I. Alpha1-chimaerin, a Rac1 GTPase-activating protein, is expressed at reduced mRNA levels in the brain of Alzheimer’s disease patients. Neurosci. Lett. 2015, 591, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Michael, S.; Xu, T.; Sarkeshik, A.; Moresco, J.J.; Yates, J.R.; Masliah, E.; Bokoch, G.M.; DerMardirossian, C. A novel Rac1 GAP splice variant relays poly-Ub accumulation signals to mediate Rac1 inactivation. Mol. Biol. Cell 2013, 24, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.; Jeoung, M.; Koo, Y.; Ji, H.; Markesbery, W.R.; Ji, I.; Ji, T.H. Kalirin is under-expressed in Alzheimer’s disease hippocampus. J. Alzheimer’s Dis. 2007, 11, 385–397. [Google Scholar] [CrossRef]
- Youn, H.; Ji, I.; Ji, H.P.; Markesbery, W.R.; Ji, T.H. Under-expression of Kalirin-7 Increases iNOS Activity in Cultured Cells and Correlates to Elevated iNOS Activity in Alzheimer’s Disease Hippocampus1. J. Alzheimer’s Dis. 2007, 12, 271–281. [Google Scholar] [CrossRef]
- Kuwano, R.; Miyashita, A.; Arai, H.; Asada, T.; Imagawa, M.; Shoji, M.; Higuchi, S.; Urakami, K.; Kakita, A.; Takahashi, H. Dynamin-binding protein gene on chromosome 10q is associated with late-onset Alzheimer’s disease. Hum. Mol. Genet. 2006, 15, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; de Pooter, T.; de Deyn, P.P.; Sleegers, K.; van Broeckhoven, C. DNMBP is genetically associated with Alzheimer dementia in the Belgian population. Neurobiol. Aging 2009, 30, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Osaka, H.; Sugiyama, S.; Kurosawa, K.; Mizuguchi, T.; Nishiyama, K.; Nishimura, A.; Tsurusaki, Y.; Miyake, N.; Harada, N. Early infantile epileptic encephalopathy associated with the disrupted gene encoding Slit-Robo Rho GTPase activating protein 2 (SRGAP2). Am. J. Med. Genet. Part A 2012, 158, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Chilton, J.; Psatha, M.; Cheng, L.; Andrews, C.; Chan, W.-M.; Law, K.; Crosier, M.; Lindsay, S.; Cheung, M. Human CHN1 mutations hyperactivate α2-chimaerin and cause Duane’s retraction syndrome. Science 2008, 321, 839–843. [Google Scholar] [CrossRef]
- Miyake, N.; Demer, J.L.; Shaaban, S.; Andrews, C.; Chan, W.-M.; Christiansen, S.P.; Hunter, D.G.; Engle, E.C. Expansion of the CHN1 strabismus phenotype. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6321–6328. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, J.E.; Baskaran, P.; Clark, C.; Hendry, A.; Lerner, O.; Hintze, M.; Allen, J.; Chilton, J.K.; Guthrie, S. Axon guidance in the developing ocular motor system and Duane retraction syndrome depends on Semaphorin signaling via alpha2-chimaerin. Proco. Natl. Acad. Sci. USA 2012, 109, 14669–14674. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef]
- Yang, Y.; Hentati, A.; Deng, H.-X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.-Y.; Ouahchi, K.; Yan, J.; Azim, A.C. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Gros-Louis, F.; IMeijer, A.; Hand, C.K.; Dubé, M.P.; MacGregor, D.L.; Seni, M.H.; Devon, R.S.; Hayden, M.R.; Andermann, F.; Andermann, E. An ALS2 gene mutation causes hereditary spastic paraplegia in a Pakistani kindred. Ann. Neurol. 2003, 53, 144–145. [Google Scholar] [CrossRef]
- Beutler, A.S.; Kulkarni, A.A.; Kanwar, R.; Klein, C.J.; Therneau, T.M.; Qin, R.; Banck, M.S.; Boora, G.K.; Ruddy, K.J.; Wu, Y. Sequencing of Charcot–Marie–Tooth disease genes in a toxic polyneuropathy. Ann. Neurol. 2014, 76, 727–737. [Google Scholar] [CrossRef]
- Ekenstedt, K.J.; Becker, D.; Minor, K.M.; Shelton, G.D.; Patterson, E.E.; Bley, T.; Oevermann, A.; Bilzer, T.; Leeb, T.; Drögemüller, C. An ARHGEF10 deletion is highly associated with a juvenile-onset inherited polyneuropathy in Leonberger and Saint Bernard dogs. PLoS Genet. 2014, 10, e1004635. [Google Scholar] [CrossRef]
- Verhoeven, K.; de Jonghe, P.; van de Putte, T.; Nelis, E.; Zwijsen, A.; Verpoorten, N.; de Vriendt, E.; Jacobs, A.; van Gerwen, V.; Francis, A. Slowed conduction and thin myelination of peripheral nerves associated with mutant rho Guanine-nucleotide exchange factor 10. Am. J. Hum. Genet. 2003, 73, 926–932. [Google Scholar] [CrossRef]
- Boora, G.K.; Kulkarni, A.A.; Kanwar, R.; Beyerlein, P.; Qin, R.; Banck, M.S.; Ruddy, K.J.; Pleticha, J.; Lynch, C.A.; Behrens, R.J. Association of the Charcot–Marie–Tooth disease gene ARHGEF10 with paclitaxel induced peripheral neuropathy in NCCTG N08CA (Alliance). J. Neurol. Sci. 2015, 357, 35–40. [Google Scholar] [CrossRef]
- Ishikawa, K.; Toru, S.; Tsunemi, T.; Li, M.; Kobayashi, K.; Yokota, T.; Amino, T.; Owada, K.; Fujigasaki, H.; Sakamoto, M. An autosomal dominant cerebellar ataxia linked to chromosome 16q22. 1 is associated with a single-nucleotide substitution in the 5′ untranslated region of the gene encoding a protein with spectrin repeat and Rho guanine-nucleotide exchange-factor domains. Am. J. Hum. Genet. 2005, 77, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Zubeldia-Brenner, L.; Gutierrez-Uzquiza, A.; Barrio-Real, L.; Wang, H.; Kazanietz, M.G.; Leskow, F.C. β3-Chimaerin, a novel member of the chimaerin Rac-GAP family. Mol. Biol. Rep. 2014, 41, 2067–2076. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Liu, S.; Zhang, J.; Zhang, Y. New member of the guanosine triphosphatase activating protein family in the human epididymis. Acta Biochim. Biophys. Sin. 2008, 40, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.-Y.C.; Taylor, S.H.; Garva, R.; Holmes, D.F.; Zeef, L.A.; Soininen, R.; Boot-Handford, R.P.; Kadler, K.E. Arhgap28 is a RhoGAP that inactivates RhoA and downregulates stress fibers. PLoS ONE 2014, 9, e107036. [Google Scholar] [CrossRef]
- Moon, S.Y.; Zang, H.; Zheng, Y. Characterization of a brain-specific Rho GTPase-activating protein, p200RhoGAP. J. Biol. Chem. 2003, 278, 4151–4159. [Google Scholar] [CrossRef]
- Nasu-Nishimura, Y.; Hayashi, T.; Ohishi, T.; Okabe, T.; Ohwada, S.; Hasegawa, Y.; Senda, T.; Toyoshima, C.; Nakamura, T.; Akiyama, T. Role of the Rho GTPase-activating protein RICS in neurite outgrowth. Genes Cell 2006, 11, 607–614. [Google Scholar] [CrossRef]
- Vo, N.; Klein, M.E.; Varlamova, O.; Keller, D.M.; Yamamoto, T.; Goodman, R.H.; Impey, S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 16426–16431. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Nuttle, X.; Sudmant, P.H.; Antonacci, F.; Graves, T.A.; Nefedov, M.; Rosenfeld, J.A.; Sajjadian, S.; Malig, M.; Kotkiewicz, H. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 2012, 149, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Charrier, C.; Josh, K.; Coutinho-Budd, J.J.; Kim, J.E.; Lambert, N.; de Marchena, J.; Jin, W.L.; Vanderhaeghen, P.; Ghosh, A.; Sassa, T.; et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 2012, 149, 923–935. [Google Scholar] [CrossRef]
- Chiang, S.H.; Hwang, J.; Legendre, M.; Zhang, M.; Kimura, A.; Saltiel, A.R. TCGAP, a multidomain Rho GTPase-activating protein involved in insulin-stimulated glucose transport. EMBO J. 2003, 22, 2679–2691. [Google Scholar] [CrossRef]
- Liu, H.; Nakazawa, T.; Tezuka, T.; Yamamoto, T. Physical and functional interaction of Fyn tyrosine kinase with a brain-enriched Rho GTPase-activating protein TCGAP. J. Biol. Chem. 2006, 281, 23611–23619. [Google Scholar] [CrossRef] [PubMed]
- Lipska, B.K.; Weinberger, D.R. To model a psychiatric disorder in animals: Schizophrenia as a reality test. Neuropsychopharmacology 2000, 23, 223–239. [Google Scholar] [CrossRef]
- Williams, L.E.; Blackford, J.U.; Luksik, A.; Gauthier, I.; Heckers, S. Reduced habituation in patients with schizophrenia. Schizophr. Res. 2013, 151, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Schalock, R.L.; Luckasson, R.A.; Shogren, K.A. The renaming of mental retardation: Understanding the change to the term intellectual disability. Intellect. Dev. Disabil. 2007, 45, 116–124. [Google Scholar] [CrossRef]
- Cicchetti, P.; Ridley, A.; Zheng, Y.; Cerione, R.; Baltimore, D. 3BP-1, an SH3 domain binding protein, has GAP activity for Rac and inhibits growth factor-induced membrane ruffling in fibroblasts. EMBO J. 1995, 14, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Janossy, A.; Dahl, J.; Bertaso, F.; Perroy, J.; Varrault, A.; Vidal, M.; Worley, P.F.; Boeckers, T.M.; Bockaert, J. Shank3-Rich2 interaction regulates AMPA receptor recycling and synaptic long-term potentiation. J. Neurosci. 2013, 33, 9699–9715. [Google Scholar] [CrossRef]
- Krugmann, S.; Anderson, K.; Ridley, S.; Risso, N.; McGregor, A.; Coadwell, J.; Davidson, K.; Eguinoa, A.; Ellson, C.; Lipp, P. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol. Cell 2002, 9, 95–108. [Google Scholar] [CrossRef]
- Miura, K.; Jacques, K.M.; Stauffer, S.; Kubosaki, A.; Zhu, K.; Hirsch, D.S.; Resau, J.; Zheng, Y.; Randazzo, P.A. ARAP1: A point of convergence for Arf and Rho signaling. Mol. Cell 2002, 9, 109–119. [Google Scholar] [CrossRef]
- De Kreuk, B.-J.; Schaefer, A.; Anthony, E.C.; Tol, S.; Fernandez-Borja, M.; Geerts, D.; Pool, J.; Hambach, L.; Goulmy, E.; Hordijk, P.L. The human minor histocompatibility antigen1 is a RhoGAP. PLoS ONE 2013, 8, e73962. [Google Scholar] [CrossRef] [PubMed]
- Spierings, E.; Wieles, B.; Goulmy, E. Minor histocompatibility antigens–big in tumour therapy. Trends Immunol. 2004, 25, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Aresta, S.; Béranger, F.; de Gunzburg, J. A novel Rho GTPase-activating-protein interacts with Gem, a member of the Ras superfamily of GTPases. Biochem. J. 2002, 367, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Saras, J.; Franzén, P.; Aspenström, P.; Hellman, U.; Gonez, L.J.; Heldin, C.-H. A novel GTPase-activating protein for Rho interacts with a PDZ domain of the protein-tyrosine phosphatase PTPL1. J. Biol. Chem. 1997, 272, 24333–24338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, C.; Wang, S.; Huang, W.; Zhou, Z.; Ying, K.; Xie, Y.; Mao, Y. Cloning and characterization of ARHGAP12, a novel human rhoGAP gene. Int. J. Biochem. Cell Biol. 2002, 34, 325–331. [Google Scholar] [CrossRef]
- Ota, H.; Hikita, T.; Sawada, M.; Nishioka, T.; Matsumoto, M.; Komura, M.; Ohno, A.; Kamiya, Y.; Miyamoto, T.; Asai, N. Speed control for neuronal migration in the postnatal brain by Gmip-mediated local inactivation of RhoA. Nat. Commun. 2014, 5, 4532. [Google Scholar] [CrossRef]
- Van den Boom, F.; Düssmann, H.; Uhlenbrock, K.; Abouhamed, M.; Bähler, M. The Myosin IXb motor activity targets the myosin IXb RhoGAP domain as cargo to sites of actin polymerization. Mol. Biol. Cell 2007, 18, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Mott, H.R.; Owen, D. Structure and Function of RLIP76 (RalBP1): An Intersection Point between Ras and Rho Signalling; Portland Press Limited: London, UK, 2014. [Google Scholar]
- Goldfinger, L.E.; Ptak, C.; Jeffery, E.D.; Shabanowitz, J.; Hunt, D.F.; Ginsberg, M.H. RLIP76 (RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J. Cell Biol. 2006, 174, 877–888. [Google Scholar] [CrossRef]
- Oby, E.; Janigro, D. The blood–brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef]
- Luo, T.; Roman, P.; Liu, C.; Sun, X.; Park, Y.; Hu, B. Upregulation of the GEF-H1 pathway after transient cerebral ischemia. Exp. Neurol. 2015, 263, 306–313. [Google Scholar] [CrossRef]
- Yokota, T.; Kouno, J.; Adachi, K.; Takahashi, H.; Teramoto, A.; Matsumoto, K.; Sugisaki, Y.; Onda, M.; Tsunoda, T. Identification of histological markers for malignant glioma by genome-wide expression analysis: Dynein, α-PIX and sorcin. Acta Neuropathol. 2006, 111, 29–38. [Google Scholar] [CrossRef]
- Nodé-Langlois, R.; Muller, D.; Boda, B. Sequential implication of the mental retardation proteins ARHGEF6 and PAK3 in spine morphogenesis. J. Cell Sci. 2006, 119, 4986–4993. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.-M.; Harvey, R.J.; Schwarz, G. Gephyrin: Where do we stand, where do we go? Trends Neurosci. 2008, 31, 257–264. [Google Scholar] [CrossRef]
- Ma, X.M.; Huang, J.P.; Eipper, B.A.; Mains, R.E. Expression of Trio, a member of the Dbl family of Rho GEFs in the developing rat brain. J. Comp. Neurol. 2005, 482, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Liu, S.; Wang, X.; Zhang, J.; Zhang, T.; Liu, Z.; Wang, D.; Zhang, A.; Zhu, M.; Gao, J. Trio gene is required for mouse learning ability. Brain Res. 2015, 1608, 82–90. [Google Scholar] [CrossRef]
- Iyer, S.C.; Wang, D.; Iyer, E.P.R.; Trunnell, S.A.; Meduri, R.; Shinwari, R.; Sulkowski, M.J.; Cox, D.N. The RhoGEF trio functions in sculpting class specific dendrite morphogenesis in Drosophila sensory neurons. PLoS ONE 2012, 7, e33634. [Google Scholar] [CrossRef]
- Miller, M.B.; Yan, Y.; Eipper, B.A.; Mains, R.E. Neuronal Rho GEFs in synaptic physiology and behavior. Neuroscientist 2013, 19, 255–273. [Google Scholar] [CrossRef]
- Shivalkar, M.; Giniger, E. Control of dendritic morphogenesis by Trio in Drosophila melanogaster. PLoS ONE 2012, 7, e33737. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Schmidt, S.; Dietzl, G.; Keleman, K.; Åsling, B.; Debant, A.; Dickson, B.J. Trio combines with dock to regulate Pak activity during photoreceptor axon pathfinding in Drosophila. Cell 2000, 101, 283–294. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.; Tian, C.; Arnesano, C.; Katritch, V.; Herring, B.E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commmun. 2017, 8, 601. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Beeser, A.; Chernoff, J.; Schiller, M.R.; Eipper, B.A.; Mains, R.E.; Huganir, R.L. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 2003, 37, 263–274. [Google Scholar] [CrossRef]
- Edvardson, S.; Wang, H.; Dor, T.; Atawneh, O.; Yaacov, B.; Gartner, J.; Cinnamon, Y.; Chen, S.; Elpeleg, O. Microcephaly-dystonia due to mutated PLEKHG2 with impaired actin polymerization. Neurogenetics 2016, 17, 25–30. [Google Scholar] [CrossRef]
- Gupta, M.; Kamynina, E.; Morley, S.; Chung, S.; Muakkassa, N.; Wang, H.; Brathwaite, S.; Sharma, G.; Manor, D. Plekhg4 is a novel Dbl family guanine nucleotide exchange factor protein for rho family GTPases. J. Biol. Chem. 2013, 288, 14522–14530. [Google Scholar] [CrossRef]
- Ge, Y.; Li, N.; Wang, Z.; Wang, J.; Cai, H. Novel variant in the FGD1 gene causing Aarskog-Scott syndrome. Exp. Med. 2017, 13, 2623–2628. [Google Scholar] [CrossRef]
- Zheng, Y.; Fischer, D.J.; Santos, M.F.; Tigyi, G.; Pasteris, N.G.; Gorski, J.L.; Xu, Y. The faciogenital dysplasia gene product FGD1 functions as a Cdc42Hs-specific guanine-nucleotide exchange factor. J. Biol. Chem. 1996, 271, 33169–33172. [Google Scholar] [CrossRef]
- Bhavsar, P.J.; Vigorito, E.; Turner, M.; Ridley, A.J. Vav GEFs regulate macrophage morphology and adhesion-induced Rac and Rho activation. Exp. Cell Res. 2009, 315, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Jagodic, M.; Colacios, C.; Nohra, R.; Dejean, A.S.; Beyeen, A.D.; Khademi, M.; Casemayou, A.; Lamouroux, L.; Duthoit, C.; Papapietro, O. A role for VAV1 in experimental autoimmune encephalomyelitis and multiple sclerosis. Sci. Transl. Med. 2009, 1, 10ra21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ligands | Receptors | GAPs | GEFs | References |
|---|---|---|---|---|
| Netrin-1 | DCC | - | Trio DOCK180 | [84,85,86] |
| Ephrin | Eph | α2-chimaerin | VAV2 and 3 Ephexin1 | [67,87,88,89,90,91,92,93] |
| Semaphorin | Plexin/Neuropilin | p190RhoGAP | PDZ-RhoGEF LARG FARP2 | [60,61,94,95] |
| Slit | Robo | vilse/CrGAP SrGAP | Sos | [63,64,96,97,98,99] |
| Diseases and Disorders | GAPs | GEFs | Studied Organisms | References |
|---|---|---|---|---|
| Bipolar disorder | ARAP1, ARAP3, ARHGAP12, ARHGAP29, ARHGAP40, ARHGAP45 | Human | [110] | |
| Cognitive complications | ARHGAP15, 2-chimaerin | ARHGEF6, Trio, Kalirin | Mouse | [111,112,113,114,115,116,117] |
| Schizophrenia | ARHGAP18, ARHGAP33, Myosin IXb | Kalirin Trio VAV3 | Human | [115,118,119,120,121,122,123,124,125,126] |
| Migraine | ARHGAP28 | Human | [127] | |
| Huntington’s disease | ARHGAP32 | GEF-H1 | Mouse, Rat | [128,129] |
| Autism spectrum disorders (ASD) | ARHGAP33, ARHGAP44 | AKAP13, Collybistin, Trio | Human, Mouse | [121,122,130,131,132,133,134,135,136] |
| Depressive disorders | GMIP | Kalirin | Human, Rat, Mouse | [114,115,116,117,137] |
| Intellectual disability | OPHN1 srGAP3 | GEF-H1, ARHGEF6, FGD1, Collybistin, Trio ARHGEF42 | Human, Mouse | [131,136,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158] |
| Seizure | RalBP1 | Collybistin | Human, Mouse | [131,149,150,151,152,153,159,160] |
| Alzheimer’s disease | SH3BP1, α1-chimaerin | ARHGEF36, Kalirin | Human | [161,162,163,164,165,166] |
| Infantile epileptic encephalopathy | srGAP2 | Human | [167] | |
| Duane’s retraction syndrome | 2-chimaerin | Human | [168,169,170] | |
| Amyotrophic lateral sclerosis | Alsin | Human | [171,172,173] | |
| Epilepsy | OPHN1 | Collybistin, Kalirin | Human, Rat | [114,115,116,117,131,143,144,145,149,150,151,152,153] |
| Charcot–Marie–Tooth disease and polyneuropathy | ARHGEF10 | Human, Dog | [174,175,176,177] | |
| Attention deficit hyperactivity disorder | Kalirin | Human | [114,115,116,117] | |
| Cocaine addiction | Kalirin | Mouse | [117] | |
| Cerebellar ataxia | PLEKHG4 | Human | [178] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niftullayev, S.; Lamarche-Vane, N. Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guidance and Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 1497. https://doi.org/10.3390/ijms20061497
Niftullayev S, Lamarche-Vane N. Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guidance and Neurological Disorders. International Journal of Molecular Sciences. 2019; 20(6):1497. https://doi.org/10.3390/ijms20061497
Chicago/Turabian StyleNiftullayev, Sadig, and Nathalie Lamarche-Vane. 2019. "Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guidance and Neurological Disorders" International Journal of Molecular Sciences 20, no. 6: 1497. https://doi.org/10.3390/ijms20061497
APA StyleNiftullayev, S., & Lamarche-Vane, N. (2019). Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guidance and Neurological Disorders. International Journal of Molecular Sciences, 20(6), 1497. https://doi.org/10.3390/ijms20061497