Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review


Abstract
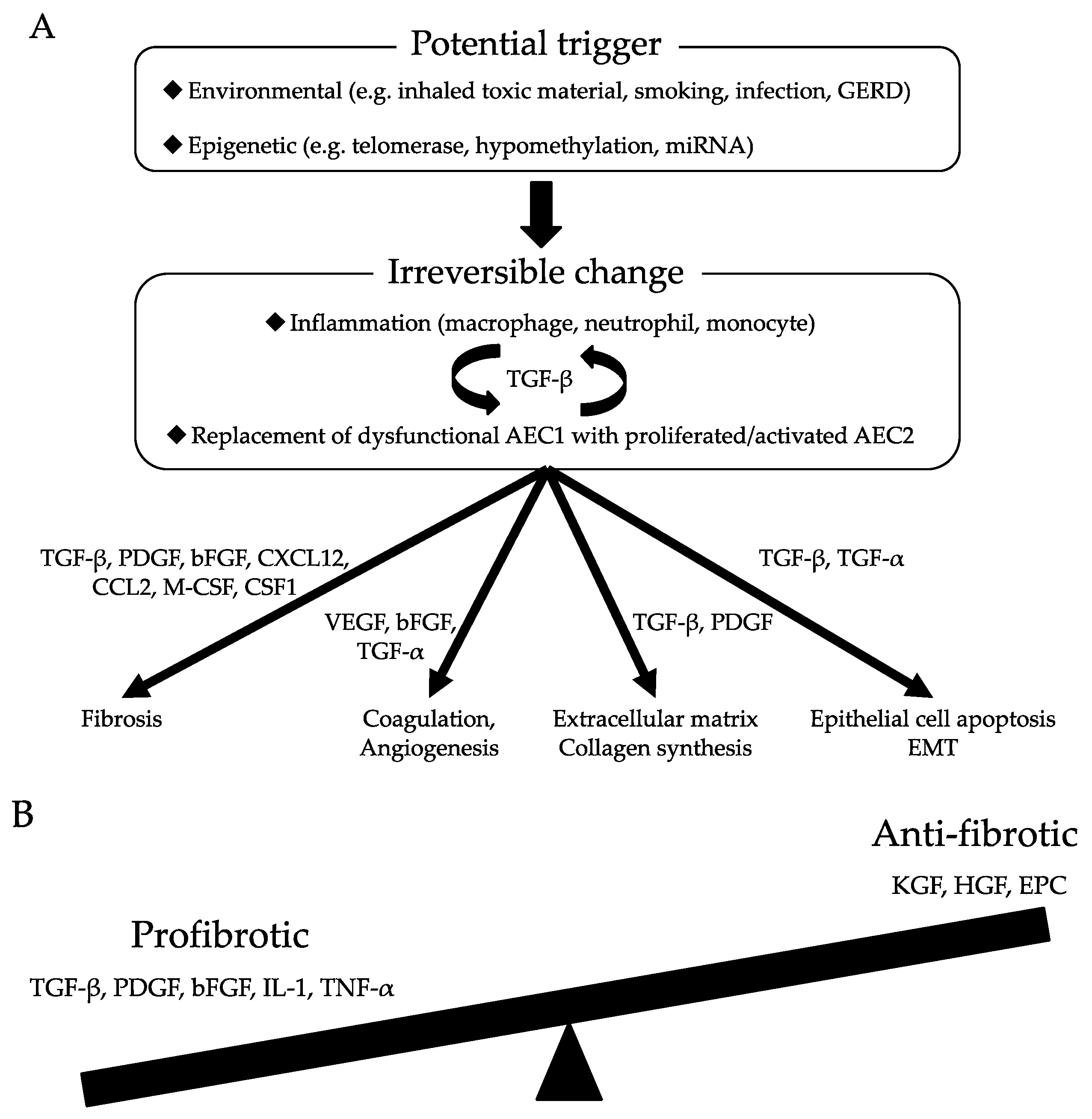
1. Introduction
2. The Pathogenesis of Pulmonary Fibrosis
2.1. Dysfunctional Epithelia Trigger Aberrant Wound Healing Processes
2.2. Growth Factors Associated with the Initial Stages of Pulmonary Fibrogenesis
2.2.1. TGF-β
2.2.2. PDGF
2.2.3. FGF
2.2.4. TGF-α
2.2.5. Keratinocyte Growth Factor (KGF)
2.2.6. Hepatocyte Growth Factor (HGF)
2.3. Changes in AEC2s that Lead to Aberrant Tissue Repair
2.3.1. UPR
2.3.2. Epithelial–Mesenchymal Transition (EMT)
2.3.3. Wnt-β-Catenin Signaling
2.4. Endothelium and Coagulation
2.5. Immunogenic Changes that Lead to Pulmonary Fibrosis
2.6. Interactions Between ECM and Mesenchymal Cells, Fibrocytes, Fibroblasts, and Myofibroblasts
3. Common Characteristics of IPF and Lung Cancer
3.1. Epigenetic and Genetic Abnormalities
3.2. Abnormal Cell–Cell Communication
3.3. Abnormal Activation of Signaling Pathways
3.4. Abnormal Migration and Invasion Activities
3.5. Inflammatory Environment
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ats/ers/jrs/alat statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Yoshida, S.; Fujiwara, T.; Wada, H.; Nakajima, T.; Suzuki, H.; Yoshino, I. Effect of Perioperative Pirfenidone Treatment in Lung Cancer Patients With Idiopathic Pulmonary Fibrosis. Ann. Thorac. Surg. 2016, 102, 1905–1910. [Google Scholar] [CrossRef]
- Iwata, T.; Yoshida, S.; Nagato, K.; Nakajima, T.; Suzuki, H.; Tagawa, T.; Mizobuchi, T.; Ota, S.; Nakatani, Y.; Yoshino, I. Experience with perioperative pirfenidone for lung cancer surgery in patients with idiopathic pulmonary fibrosis. Surg. Today 2015, 45, 1263–1270. [Google Scholar] [CrossRef]
- Iwata, T.; Yoshino, I.; Yoshida, S.; Ikeda, N.; Tsuboi, M.; Asato, Y.; Katakami, N.; Sakamoto, K.; Yamashita, Y.; Okami, J.; et al. A phase II trial evaluating the efficacy and safety of perioperative pirfenidone for prevention of acute exacerbation of idiopathic pulmonary fibrosis in lung cancer patients undergoing pulmonary resection: West Japan Oncology Group 6711 L (PEOPLE Study). Respir. Res. 2016, 17, 90. [Google Scholar] [CrossRef]
- Costabel, U.; Albera, C.; Lancaster, L.H.; Lin, C.Y.; Hormel, P.; Hulter, H.N.; Noble, P.W. An Open-Label Study of the Long-Term Safety of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (RECAP). Respiration 2017, 94, 408–415. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef]
- Goto, T.; Maeshima, A.; Akanabe, K.; Oyamada, Y.; Kato, R. Acute exacerbation of idiopathic pulmonary fibrosis of microscopic usual interstitial pneumonia pattern after lung cancer surgery. Ann. Thorac. Cardiovasc. Surg. 2011, 17, 573–576. [Google Scholar] [CrossRef]
- Goto, T.; Maeshima, A.; Oyamada, Y.; Kato, R. Idiopathic pulmonary fibrosis as a prognostic factor in non-small cell lung cancer. Int. J. Clin. Oncol. 2014, 19, 266–273. [Google Scholar] [CrossRef]
- Goto, T. Measuring Surgery Outcomes of Lung Cancer Patients with Concomitant Pulmonary Fibrosis: A Review of the Literature. Cancers 2018, 10, 223. [Google Scholar] [CrossRef]
- Hendriks, L.E.; Drent, M.; van Haren, E.H.; Verschakelen, J.A.; Verleden, G.M. Lung cancer in idiopathic pulmonary fibrosis patients diagnosed during or after lung transplantation. Respir. Med. Case Rep. 2012, 5, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.E.; Jett, J.R. Does interstitial lung disease predispose to lung cancer? Curr. Opin. Pulm. Med. 2005, 11, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Nepali, K.; Liou, J.P. Idiopathic Pulmonary Fibrosis: Current Status, Recent Progress, and Emerging Targets. J. Med. Chem. 2017, 60, 527–553. [Google Scholar] [CrossRef] [PubMed]
- Betensley, A.; Sharif, R.; Karamichos, D. A Systematic Review of the Role of Dysfunctional Wound Healing in the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.K.; Laurent, G.J.; Shahzeidi, S.; Lympany, P.A.; du Bois, R.M.; Jeffery, P.K.; McAnulty, R.J. Transforming growth factors-beta(1), -beta(2), and -beta(3) stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997, 150, 981–991. [Google Scholar]
- Khalil, N.; O’Connor, R.N.; Unruh, H.W.; Warren, P.W.; Flanders, K.C.; Kemp, A.; Bereznay, O.H.; Greenberg, A.H. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1991, 5, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Raghow, B.; Irish, P.; Kang, A.H. Coordinate regulation of transforming growth factor beta gene expression and cell proliferation in hamster lungs undergoing bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 1989, 84, 1836–1842. [Google Scholar] [CrossRef]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, H.N.; Bravo, M.A.; Avila, R.E.; Galanopoulos, T.; Neville-Golden, J.; Maxwell, M.; Selman, M. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J. Clin. Investig. 1990, 86, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Martinet, Y.; Rom, W.N.; Grotendorst, G.R.; Martin, G.R.; Crystal, R.G. Exaggerated spontaneous release of platelet-derived growth factor by alveolar macrophages from patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 1987, 317, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Morris, G.F.; Lei, W.H.; Hart, C.E.; Lasky, J.A.; Brody, A.R. Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am. J. Respir. Cell Mol. Biol. 1997, 17, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, G.W.; Li, J.; Finkelstein, J.B.; Eisenberg, T.; Liu, J.Y.; Lasky, J.A.; Athas, G.; Morris, G.F.; Brody, A.R. Emphysematous lesions, inflammation, and fibrosis in the lungs of transgenic mice overexpressing platelet-derived growth factor. Am. J. Pathol. 1999, 154, 1763–1775. [Google Scholar] [CrossRef]
- Yi, E.S.; Lee, H.; Yin, S.; Piguet, P.; Sarosi, I.; Kaufmann, S.; Tarpley, J.; Wang, N.S.; Ulich, T.R. Platelet-derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am. J. Pathol. 1996, 149, 539–548. [Google Scholar]
- Yoshida, M.; Sakuma-Mochizuki, J.; Abe, K.; Arai, T.; Mori, M.; Goya, S.; Matsuoka, H.; Hayashi, S.; Kaneda, Y.; Kishimoto, T. In vivo gene transfer of an extracellular domain of platelet-derived growth factor beta receptor by the HVJ-liposome method ameliorates bleomycin-induced pulmonary fibrosis. Biochem. Biophys. Res. Commun. 1999, 265, 503–508. [Google Scholar] [CrossRef]
- Rom, W.N.; Basset, P.; Fells, G.A.; Nukiwa, T.; Trapnell, B.C.; Crysal, R.G. Alveolar macrophages release an insulin-like growth factor I-type molecule. J. Clin. Investig. 1988, 82, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, P.B.; Adelberg, S.; Crystal, R.G. Mechanisms of pulmonary fibrosis. Spontaneous release of the alveolar macrophage-derived growth factor in the interstitial lung disorders. J. Clin. Investig. 1983, 72, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Henke, C.; Marineili, W.; Jessurun, J.; Fox, J.; Harms, D.; Peterson, M.; Chiang, L.; Doran, P. Macrophage production of basic fibroblast growth factor in the fibroproliferative disorder of alveolar fibrosis after lung injury. Am. J. Pathol. 1993, 143, 1189–1199. [Google Scholar]
- Inoue, Y.; King, T.E., Jr.; Tinkle, S.S.; Dockstader, K.; Newman, L.S. Human mast cell basic fibroblast growth factor in pulmonary fibrotic disorders. Am. J. Pathol. 1996, 149, 2037–2054. [Google Scholar] [PubMed]
- Liu, J.Y.; Morris, G.F.; Lei, W.H.; Corti, M.; Brody, A.R. Up-regulated expression of transforming growth factor-alpha in the bronchiolar-alveolar duct regions of asbestos-exposed rats. Am. J. Pathol. 1996, 149, 205–217. [Google Scholar] [PubMed]
- Madtes, D.K.; Busby, H.K.; Strandjord, T.P.; Clark, J.G. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am. J. Respir. Cell Mol. Biol. 1994, 11, 540–551. [Google Scholar] [CrossRef]
- Korfhagen, T.R.; Swantz, R.J.; Wert, S.E.; McCarty, J.M.; Kerlakian, C.B.; Glasser, S.W.; Whitsett, J.A. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J. Clin. Investig. 1994, 93, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Iyama, K.; Kuroda, M.J.; Sano, K. Double intratracheal instillation of keratinocyte growth factor prevents bleomycin-induced lung fibrosis in rats. J. Pathol. 1998, 186, 90–98. [Google Scholar] [CrossRef]
- Sakai, T.; Satoh, K.; Matsushima, K.; Shindo, S.; Abe, S.; Abe, T.; Motomiya, M.; Kawamoto, T.; Kawabata, Y.; Nakamura, T.; et al. Hepatocyte growth factor in bronchoalveolar lavage fluids and cells in patients with inflammatory chest diseases of the lower respiratory tract: Detection by RIA and in situ hybridization. Am. J. Respir. Cell Mol. Biol. 1997, 16, 388–397. [Google Scholar] [CrossRef]
- Maeda, J.; Ueki, N.; Hada, T.; Higashino, K. Elevated serum hepatocyte growth factor/scatter factor levels in inflammatory lung disease. Am. J. Respir. Crit. Care Med. 1995, 152, 1587–1591. [Google Scholar] [CrossRef]
- Shiratori, M.; Michalopoulos, G.; Shinozuka, H.; Singh, G.; Ogasawara, H.; Katyal, S.L. Hepatocyte growth factor stimulates DNA synthesis in alveolar epithelial type II cells in vitro. Am. J. Respir. Cell Mol. Biol. 1995, 12, 171–180. [Google Scholar] [CrossRef]
- Yaekashiwa, M.; Nakayama, S.; Ohnuma, K.; Sakai, T.; Abe, T.; Satoh, K.; Matsumoto, K.; Nakamura, T.; Takahashi, T.; Nukiwa, T. Simultaneous or delayed administration of hepatocyte growth factor equally represses the fibrotic changes in murine lung injury induced by bleomycin. A morphologic study. Am. J. Respir. Crit. Care Med. 1997, 156, 1937–1944. [Google Scholar] [CrossRef]
- Nagahori, T.; Dohi, M.; Matsumoto, K.; Saitoh, K.; Honda, Z.I.; Nakamura, T.; Yamamoto, K. Interferon-gamma upregulates the c-Met/hepatocyte growth factor receptor expression in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Malli, F.; Koutsokera, A.; Paraskeva, E.; Zakynthinos, E.; Papagianni, M.; Makris, D.; Tsilioni, I.; Molyvdas, P.A.; Gourgoulianis, K.I.; Daniil, Z. Endothelial progenitor cells in the pathogenesis of idiopathic pulmonary fibrosis: An evolving concept. PLoS ONE 2013, 8, e53658. [Google Scholar] [CrossRef] [PubMed]
- Zolak, J.S.; de Andrade, J.A. Idiopathic pulmonary fibrosis. Immunol. Allergy Clin. N. Am. 2012, 32, 473–485. [Google Scholar] [CrossRef]
- Kotsianidis, I.; Nakou, E.; Bouchliou, I.; Tzouvelekis, A.; Spanoudakis, E.; Steiropoulos, P.; Sotiriou, I.; Aidinis, V.; Margaritis, D.; Tsatalas, C.; et al. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 1121–1130. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Hinz, B. Mechanical aspects of lung fibrosis: A spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 2012, 9, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Tomassetti, S.; Tsitoura, E.; Vancheri, C. Idiopathic pulmonary fibrosis and lung cancer: A clinical and pathogenesis update. Curr. Opin. Pulm. Med. 2015, 21, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. 2013, 22, 265–272. [Google Scholar] [CrossRef]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Kumbla, P.; Hagood, J.S. Enhanced myofibroblastic differentiation and survival in Thy-1(-) lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2007, 36, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Pardo, A.; Selman, M.; Nuovo, G.J.; Tollefsbol, T.O.; Siegal, G.P.; Hagood, J.S. Thy-1 promoter hypermethylation: A novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 610–618. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Hojo, S.; Fujita, J.; Yamadori, I.; Kamei, T.; Yoshinouchi, T.; Ohtsuki, Y.; Okada, H.; Bandoh, S.; Yamaji, Y.; Takahara, J.; et al. Heterogeneous point mutations of the p53 gene in pulmonary fibrosis. Eur. Respir. J. 1998, 12, 1404–1408. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, K.; Yoshimura, A.; Gemma, A.; Mochimaru, H.; Hosoya, Y.; Kunugi, S.; Matsuda, K.; Seike, M.; Kurimoto, F.; Takenaka, K.; et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001, 61, 8527–8533. [Google Scholar] [PubMed]
- Demopoulos, K.; Arvanitis, D.A.; Vassilakis, D.A.; Siafakas, N.M.; Spandidos, D.A. MYCL1, FHIT, SPARC, p16(INK4) and TP53 genes associated to lung cancer in idiopathic pulmonary fibrosis. J. Cell. Mol. Med. 2002, 6, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chung, M.J.; Ullenbruch, M.; Yu, H.; Jin, H.; Hu, B.; Choi, Y.Y.; Ishikawa, F.; Phan, S.H. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J. Clin. Investig. 2007, 117, 3800–3809. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Casoni, G.L.; Ulivi, P.; Mercatali, L.; Chilosi, M.; Tomassetti, S.; Romagnoli, M.; Ravaglia, C.; Gurioli, C.; Gurioli, C.; Zoli, W.; et al. Increased levels of free circulating DNA in patients with idiopathic pulmonary fibrosis. Int. J. Biol. Markers 2010, 25, 229–235. [Google Scholar] [CrossRef]
- Lovat, F.; Valeri, N.; Croce, C.M. MicroRNAs in the pathogenesis of cancer. Semin Oncol. 2011, 38, 724–733. [Google Scholar] [CrossRef]
- Oak, S.R.; Murray, L.; Herath, A.; Sleeman, M.; Anderson, I.; Joshi, A.D.; Coelho, A.L.; Flaherty, K.R.; Toews, G.B.; Knight, D.; et al. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS ONE 2011, 6, e21253. [Google Scholar] [CrossRef]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in idiopathic pulmonary fibrosis. Transl. Res. 2011, 157, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Correll, K.A.; Edeen, K.E.; Redente, E.F.; Zemans, R.L.; Edelman, B.L.; Danhorn, T.; Curran-Everett, D.; Mikels-Vigdal, A.; Mason, R.J. TGF beta inhibits HGF, FGF7, and FGF10 expression in normal and IPF lung fibroblasts. Physiol. Rep. 2018, 6, e13794. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Feghali-Bostwick, C.A.; Amrani, Y.; Bradding, P. Lipoxin A4 Attenuates Constitutive and TGF-beta1-Dependent Profibrotic Activity in Human Lung Myofibroblasts. J. Immunol. 2015, 195, 2852–2860. [Google Scholar] [CrossRef] [PubMed]
- Samara, K.D.; Trachalaki, A.; Tsitoura, E.; Koutsopoulos, A.V.; Lagoudaki, E.D.; Lasithiotaki, I.; Margaritopoulos, G.; Pantelidis, P.; Bibaki, E.; Siafakas, N.M.; et al. Upregulation of citrullination pathway: From Autoimmune to Idiopathic Lung Fibrosis. Respir. Res. 2017, 18, 218. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Chanson, M.; Crespin, S. Connexins as therapeutic targets in lung disease. Expert Opin. Ther. Targets 2011, 15, 989–1002. [Google Scholar] [CrossRef]
- Mori, R.; Power, K.T.; Wang, C.M.; Martin, P.; Becker, D.L. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 2006, 119, 5193–5203. [Google Scholar] [CrossRef]
- Cesen-Cummings, K.; Fernstrom, M.J.; Malkinson, A.M.; Ruch, R.J. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis 1998, 19, 61–67. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Zhang, W.; Wang, N.Q.; Bani-Yaghoub, M.; Lin, Z.X.; Naus, C.C. Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin43. Carcinogenesis 1998, 19, 1889–1894. [Google Scholar] [CrossRef]
- Trovato-Salinaro, A.; Trovato-Salinaro, E.; Failla, M.; Mastruzzo, C.; Tomaselli, V.; Gili, E.; Crimi, N.; Condorelli, D.F.; Vancheri, C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 122. [Google Scholar] [CrossRef]
- Mazieres, J.; He, B.; You, L.; Xu, Z.; Jablons, D.M. Wnt signaling in lung cancer. Cancer Lett. 2005, 222, 1–10. [Google Scholar] [CrossRef]
- Bowley, E.; O’Gorman, D.B.; Gan, B.S. Beta-catenin signaling in fibroproliferative disease. J. Surg. Res. 2007, 138, 141–150. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Caraci, F.; Gili, E.; Calafiore, M.; Failla, M.; La Rosa, C.; Crimi, N.; Sortino, M.A.; Nicoletti, F.; Copani, A.; Vancheri, C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res. 2008, 57, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6, e24663. [Google Scholar] [CrossRef]
- Guerreiro, A.S.; Fattet, S.; Kulesza, D.W.; Atamer, A.; Elsing, A.N.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; et al. A sensitized RNA interference screen identifies a novel role for the PI3K p110gamma isoform in medulloblastoma cell proliferation and chemoresistance. Mol. Cancer Res. 2011, 9, 925–935. [Google Scholar] [CrossRef]
- Wei, X.; Han, J.; Chen, Z.Z.; Qi, B.W.; Wang, G.C.; Ma, Y.H.; Zheng, H.; Luo, Y.F.; Wei, Y.Q.; Chen, L.J. A phosphoinositide 3-kinase-gamma inhibitor, AS605240 prevents bleomycin-induced pulmonary fibrosis in rats. Biochem. Biophys. Res. Commun. 2010, 397, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Schermuly, R.T.; Ghofrani, H.A. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat. Rev. Drug Discov. 2010, 9, 956–970. [Google Scholar] [CrossRef]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Muller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 2007, 29, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Miyazaki, E.; Ito, T.; Hiroshige, S.; Nureki, S.I.; Ueno, T.; Takenaka, R.; Fukami, T.; Kumamoto, T. Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung 2010, 188, 247–252. [Google Scholar] [CrossRef]
- Rhee, C.K.; Lee, S.H.; Yoon, H.K.; Kim, S.C.; Lee, S.Y.; Kwon, S.S.; Kim, Y.K.; Kim, K.H.; Kim, T.J.; Kim, J.W. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respiration 2011, 82, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Guyot, C.; Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45 (Suppl. 2), S163–S175. [Google Scholar] [CrossRef]
- Fletcher, C.D. Myofibroblastic tumours: An update. Verh. Dtsch. Ges. Pathol. 1998, 82, 75–82. [Google Scholar] [PubMed]
- Vancheri, C.; Sortino, M.A.; Tomaselli, V.; Mastruzzo, C.; Condorelli, F.; Bellistri, G.; Pistorio, M.P.; Canonico, P.L.; Crimi, N. Different expression of TNF-alpha receptors and prostaglandin E(2)Production in normal and fibrotic lung fibroblasts: Potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell Mol. Biol. 2000, 22, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.S.; Zhou, Y.; et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef]
- Bernau, K.; Ngam, C.; Torr, E.E.; Acton, B.; Kach, J.; Dulin, N.O.; Sandbo, N. Megakaryoblastic leukemia-1 is required for the development of bleomycin-induced pulmonary fibrosis. Respir. Res. 2015, 16, 45. [Google Scholar] [CrossRef]
- Medjkane, S.; Perez-Sanchez, C.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef]
- Kim, T.; Hwang, D.; Lee, D.; Kim, J.H.; Kim, S.Y.; Lim, D.S. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017, 36, 520–535. [Google Scholar] [CrossRef]
- Kishi, T.; Mayanagi, T.; Iwabuchi, S.; Akasaka, T.; Sobue, K. Myocardin-related transcription factor A (MRTF-A) activity-dependent cell adhesion is correlated to focal adhesion kinase (FAK) activity. Oncotarget 2016, 7, 72113–72130. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Niki, T.; Yamada, T.; Matsuno, Y.; Kondo, H.; Hirohashi, S. Increased expression of laminin-5 and its prognostic significance in lung adenocarcinomas of small size. An immunohistochemical analysis of 102 cases. Cancer 2001, 91, 1129–1141. [Google Scholar] [CrossRef]
- Garrido, C.; Schmitt, E.; Cande, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. HSP27 and HSP70: Potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, G.; Pastorino, U.; Pasini, F.; Maissoneuve, P.; Fraggetta, F.; Iannucci, A.; Sonzogni, A.; De Manzoni, G.; Terzi, A.; Durante, E.; et al. Independent prognostic value of fascin immunoreactivity in stage I nonsmall cell lung cancer. Br. J. Cancer 2003, 88, 537–547. [Google Scholar] [CrossRef]
- Chilosi, M.; Zamo, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef]
- Kidera, Y.; Tsubaki, M.; Yamazoe, Y.; Shoji, K.; Nakamura, H.; Ogaki, M.; Satou, T.; Itoh, T.; Isozaki, M.; Kaneko, J.; et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 2010, 29, 127. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Behr, J.; Kolb, M.; Cox, G. Treating IPF--all or nothing? A PRO-CON debate. Respirology 2009, 14, 1072–1081. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Mills, C.D.; Lenz, L.L.; Harris, R.A. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016, 76, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Kudo-Saito, C.; Muramatsu, R.; Fujita, T.; Saito, M.; Nagumo, H.; Sakurai, T.; Noji, S.; Takahata, E.; Yaguchi, T.; et al. Determination of poor prognostic immune features of tumour microenvironment in non-smoking patients with lung adenocarcinoma. Eur. J. Cancer 2017, 86, 15–27. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Greiffo, F.R.; Frankenberger, M.; Bandres, J.; Heinzelmann, K.; Neurohr, C.; Hatz, R.; Hartl, D.; Behr, J.; Eickelberg, O. Peripheral blood myeloid-derived suppressor cells reflect disease status in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Muramatsu, R.; Fujita, T.; Nagumo, H.; Sakurai, T.; Noji, S.; Takahata, E.; Yaguchi, T.; Tsukamoto, N.; Kudo-Saito, C.; et al. Prognostic value of tumor-infiltrating lymphocytes differs depending on histological type and smoking habit in completely resected non-small-cell lung cancer. Ann. Oncol. 2016, 27, 2117–2123. [Google Scholar] [CrossRef]
- Kinoshita, T.; Ishii, G.; Hiraoka, N.; Hirayama, S.; Yamauchi, C.; Aokage, K.; Hishida, T.; Yoshida, J.; Nagai, K.; Ochiai, A. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci. 2013, 104, 409–415. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Molecules | Profibrotic/Antifibrotic | Function in IPF |
|---|---|---|
| TGF-β | Profibrotic | Extracellular matrix production |
| Epithelial mesenchymal transition | ||
| Epithelial cell apoptosis and migration | ||
| Recruitment of fibrocytes and immune cells | ||
| Fibroblast activation, myofibroblast proliferation | ||
| Induction of growth factor production | ||
| Induction of pro-angiogenic mediator production | ||
| PDGF | Profibrotic | Extracellular matrix production |
| Fibroblast proliferation | ||
| FGF | Profibrotic | Fibroblast activation |
| Endothelial cell proliferation | ||
| TGF-α | Profibrotic | Epithelial cell proliferation |
| Endothelial cell proliferation | ||
| Fibroblast proliferation | ||
| VEGF | Profibrotic | Angiogenesis in injured lung |
| KGF | Antifibrotic | Maintenance and repair of injured lung |
| HGF | Antifibrotic | Maintenance and repair of injured lung |
| Mediators | IPF | Lung Cancer | |
|---|---|---|---|
| Abnormal mRNA | let-7 | down-regulated | down-regulated |
| miR-21 | up-regulated | up-regulated | |
| miR-29 | down-regulated | down-regulated | |
| miR-30 | down-regulated | down-regulated | |
| miR-155 | up-regulated | up-regulated | |
| miR-200 | down-regulated | down-regulated | |
| Cell-free DNA | - | up-regulated | up-regulated |
| Glycoprotein | Thy-1 | down-regulated | down-regulated |
| Connexin | Cx43 | down-regulated | down-regulated |
| Growth Factors | TGF-β | up-regulated | up-regulated |
| PDGF | up-regulated | up-regulated | |
| Migration | VEGF | up-regulated | up-regulated |
| FGF | up-regulated | up-regulated | |
| laminin | up-regulated | up-regulated | |
| fascin | up-regulated | up-regulated | |
| Pathways | heat shock protein 27 | up-regulated | up-regulated |
| Wnt pathway | up-regulated | up-regulated | |
| PI3K/Akt pathway | up-regulated | up-regulated | |
| Immune Cells | FAM | up-regulated | up-regulated |
| MDSC | up-regulated | up-regulated | |
| Treg | down-regulated | up-regulated | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. https://doi.org/10.3390/ijms20061461
Kinoshita T, Goto T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. International Journal of Molecular Sciences. 2019; 20(6):1461. https://doi.org/10.3390/ijms20061461
Chicago/Turabian StyleKinoshita, Tomonari, and Taichiro Goto. 2019. "Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review" International Journal of Molecular Sciences 20, no. 6: 1461. https://doi.org/10.3390/ijms20061461
APA StyleKinoshita, T., & Goto, T. (2019). Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. International Journal of Molecular Sciences, 20(6), 1461. https://doi.org/10.3390/ijms20061461