Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer


 ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Epidemiology—Risk Factors
2. Classification—Genomic Landscapes—Drug Resistance
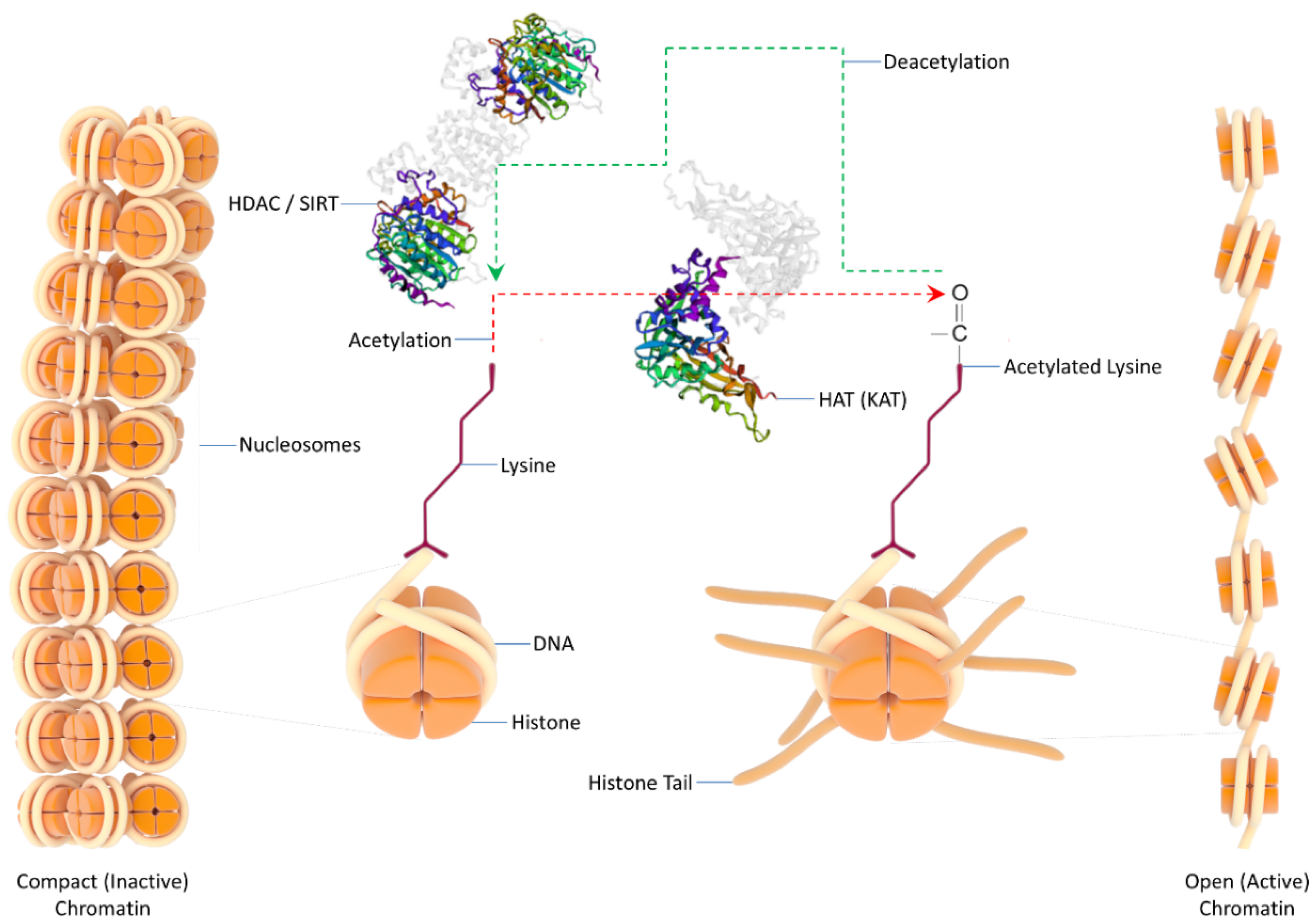
3. Chromatin Remodeling—Acetylation versus Deacetylation—HATs
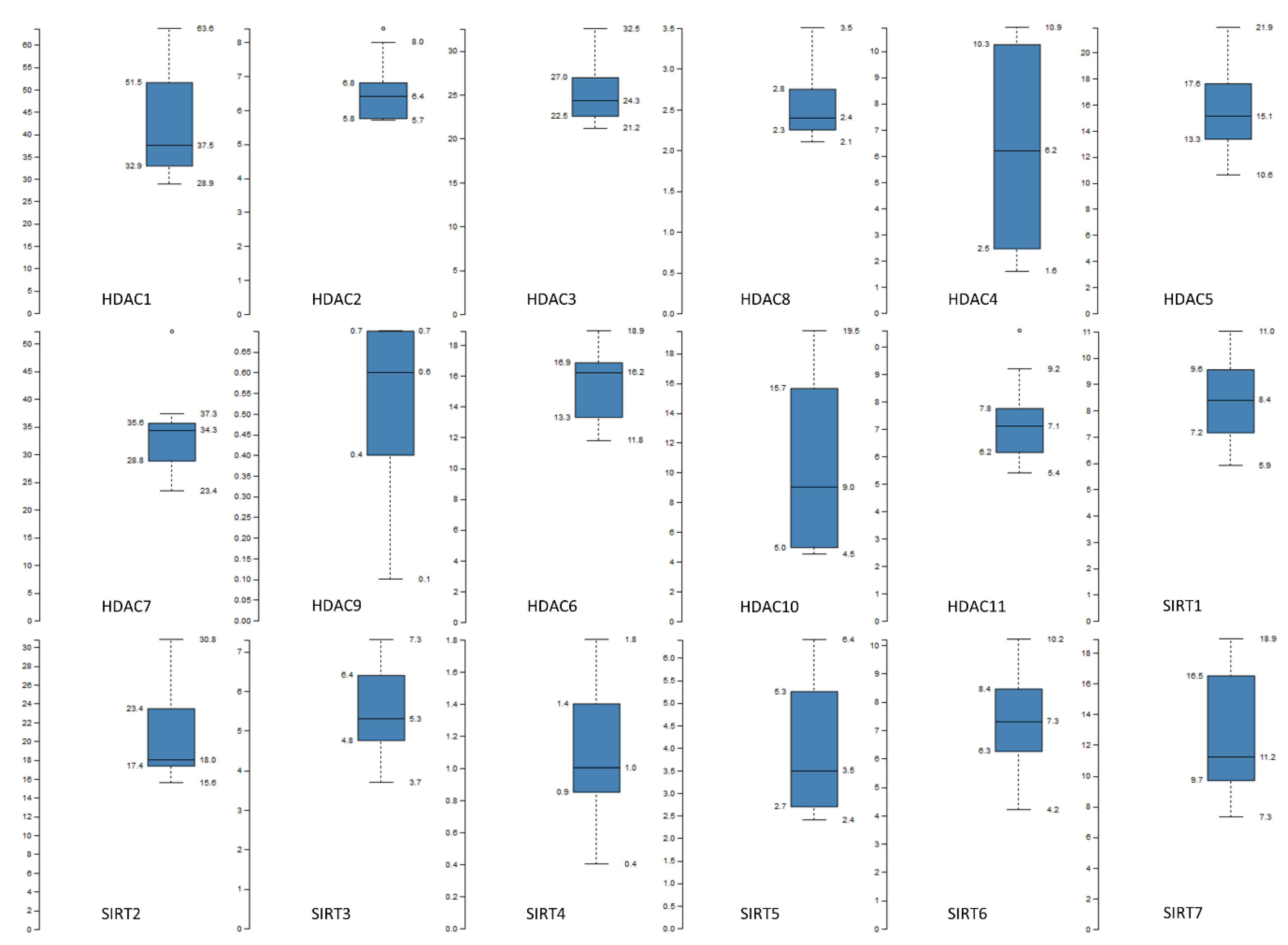
4. HDACs: Taxonomy—Activity
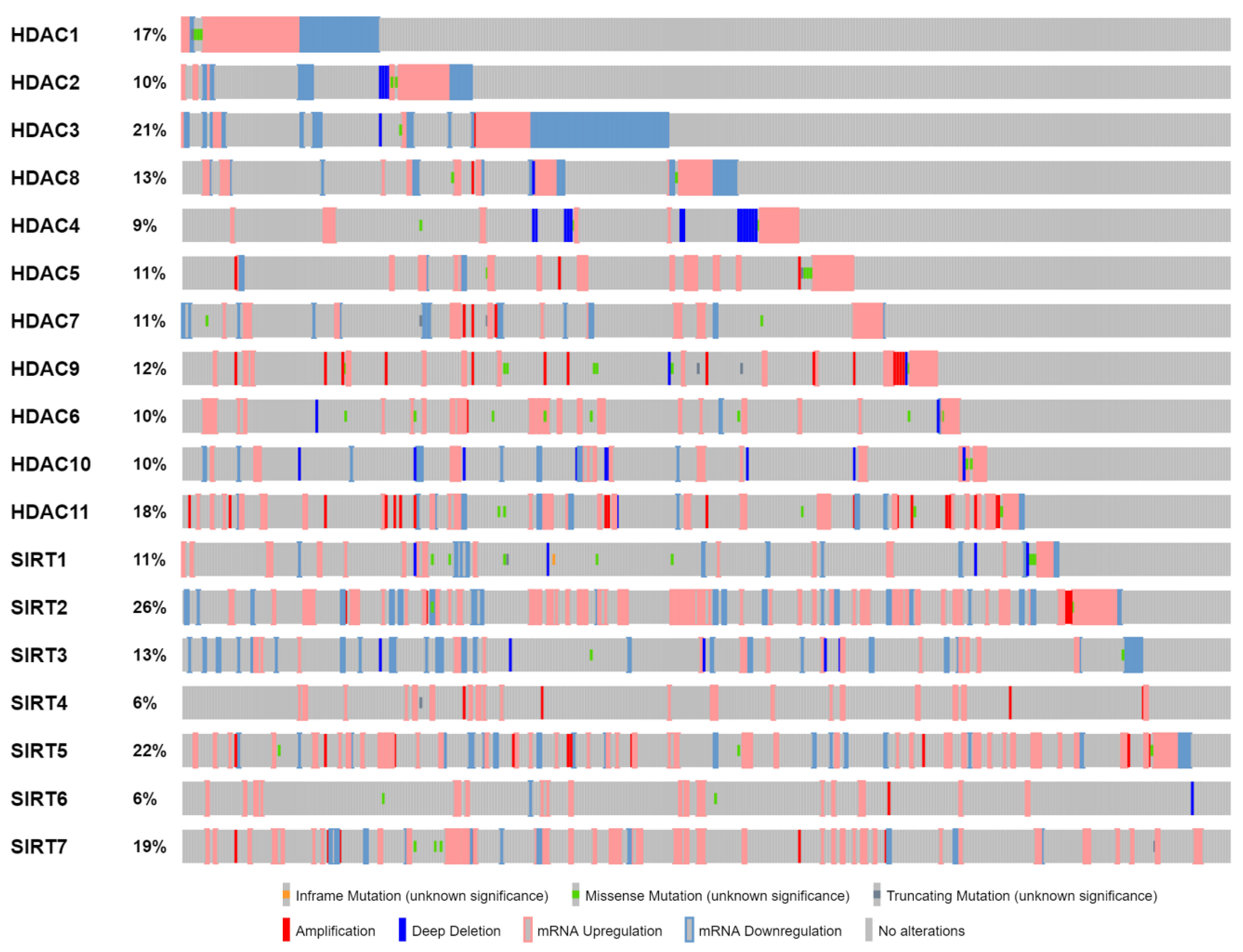
5. HDACs: Deregulation—Oncogenesis
5.1. Aberrant Recruitment
5.2. Aberrant Signaling
5.3. Aberrant Expression
6. “HDACing” Bladder Cancer
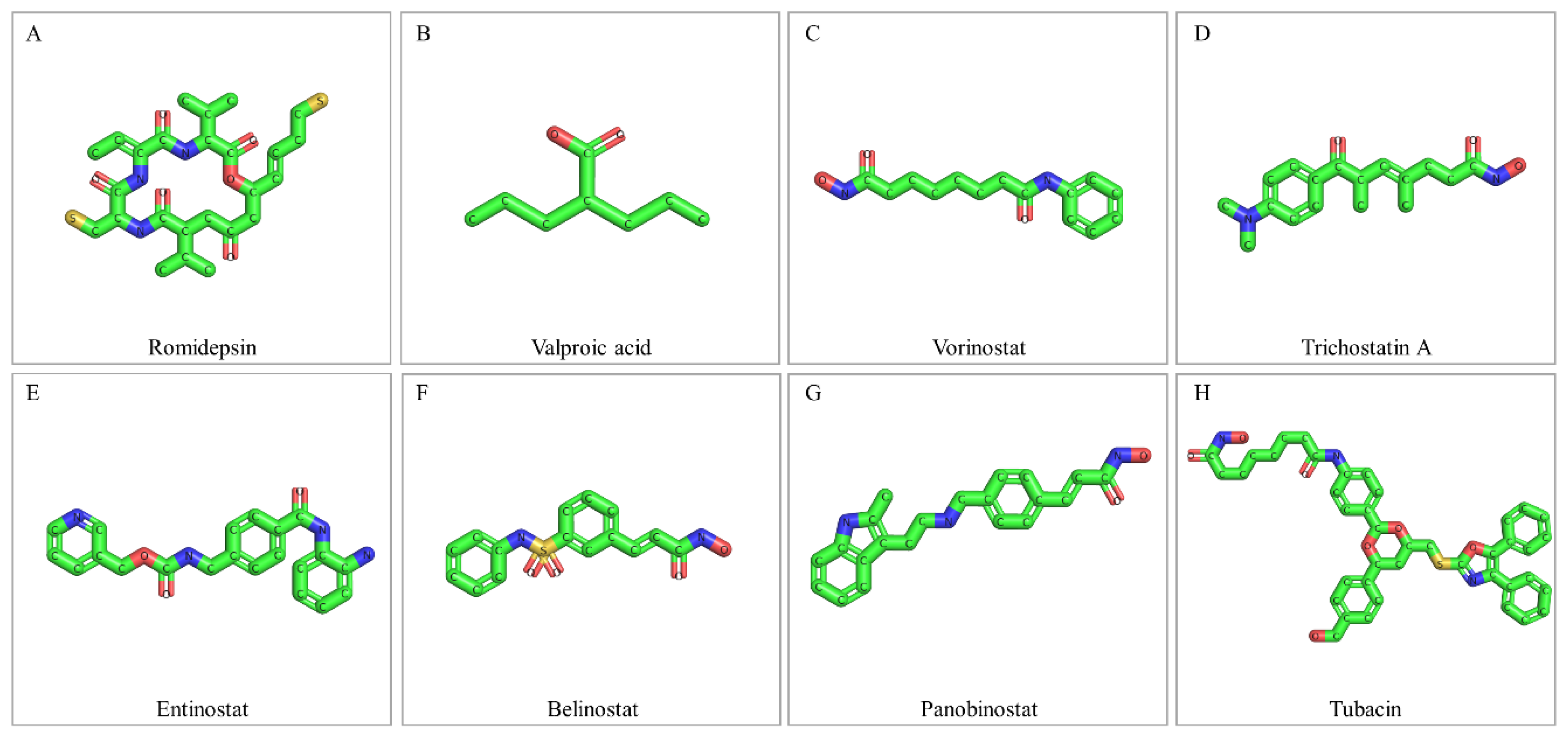
7. Drugging the Bladder Cancer “Acetyl-proteome”: HDAC Inhibitors
7.1. Romidepsin
7.2. Valproic Acid
7.3. Vorinostat
7.4. Trichostatin A
7.5. Entinostat
7.6. Belinostat
7.7. Panobinostat
7.8. Tubacin
8. Clinical Trials
Urothelial Bladder Carcinoma
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Mahdavifar, N.; Ghoncheh, M.; Pakzad, R.; Momenimovahed, Z.; Salehiniya, H. Epidemiology, Incidence and Mortality of Bladder Cancer and their Relationship with the Development Index in the World. Asian Pac. J. Cancer Prev. 2016, 17, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Kujtan, L.; Hussain, A.; Subramanian, J.; Masood, A. The evolving genomic landscape in urothelial cancer. Curr. Opin. Oncol. 2018, 30, 197–202. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.P.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017, 14, 215–229. [Google Scholar] [CrossRef]
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef]
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.S.; Ishill, N.M.; Lowrance, W.T.; Herr, H.W.; Elkin, E.B. Ethnic differences in bladder cancer survival. Urology 2011, 78, 544–549. [Google Scholar] [CrossRef]
- Cumberbatch, M.G.K.; Jubber, I.; Black, P.C.; Esperto, F.; Figueroa, J.D.; Kamat, A.M.; Kiemeney, L.; Lotan, Y.; Pang, K.; Silverman, D.T.; et al. Epidemiology of Bladder Cancer: A Systematic Review and Contemporary Update of Risk Factors in 2018. Eur. Urol. 2018, 74, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.; Marra, G.; Capoun, O.; Soukup, V.; Gontero, P. Prevention of bladder cancer incidence and recurrence: Tobacco use. Curr. Opin. Urol. 2018, 28, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Garren, B.; Nielsen, M.E.; Tang, L. Lifestyle and nutritional modifiable factors in the prevention and treatment of bladder cancer. Urol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, C.D.; Mostafid, H. Prevention of bladder cancer incidence and recurrence: Nutrition and lifestyle. Curr. Opin. Urol. 2018, 28, 88–92. [Google Scholar] [CrossRef]
- Freedman, N.D.; Silverman, D.T.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. Association between smoking and risk of bladder cancer among men and women. JAMA 2011, 306, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Hashim, D.; Boffetta, P. Occupational and environmental exposures and cancers in developing countries. Ann. Glob. Health 2014, 80, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.D.; Prasad, S.M.; Patel, A.R.; Weiner, A.B.; Pariser, J.J.; Razmaria, A.; Maene, C.; Schuble, T.; Pierce, B.; Steinberg, G.D. Bladder Cancer Mortality in the United States: A Geographic and Temporal Analysis of Socioeconomic and Environmental Factors. J. Urol. 2016, 195, 290–296. [Google Scholar] [CrossRef]
- Cumberbatch, M.G.; Windsor-Shellard, B.; Catto, J.W. The contemporary landscape of occupational bladder cancer within the United Kingdom: A meta-analysis of risks over the last 80 years. BJU Int. 2017, 119, 100–109. [Google Scholar] [CrossRef]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- An, Y.; Li, H.; Wang, K.J.; Liu, X.H.; Qiu, M.X.; Liao, Y.; Huang, J.L.; Wang, X.S. Meta-analysis of the relationship between slow acetylation of N-acetyl transferase 2 and the risk of bladder cancer. Genet. Mol. Res. 2015, 14, 16896–16904. [Google Scholar] [CrossRef]
- Rothman, N.; Garcia-Closas, M.; Chatterjee, N.; Malats, N.; Wu, X.; Figueroa, J.D.; Real, F.X.; Van Den Berg, D.; Matullo, G.; Baris, D.; et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet. 2010, 42, 978–984. [Google Scholar] [CrossRef]
- Rafnar, T.; Vermeulen, S.H.; Sulem, P.; Thorleifsson, G.; Aben, K.K.; Witjes, J.A.; Grotenhuis, A.J.; Verhaegh, G.W.; Hulsbergen-van de Kaa, C.A.; Besenbacher, S.; et al. European genome-wide association study identifies SLC14A1 as a new urinary bladder cancer susceptibility gene. Hum. Mol. Genet. 2011, 20, 4268–4281. [Google Scholar] [CrossRef]
- Garcia-Closas, M.; Ye, Y.; Rothman, N.; Figueroa, J.D.; Malats, N.; Dinney, C.P.; Chatterjee, N.; Prokunina-Olsson, L.; Wang, Z.; Lin, J.; et al. A genome-wide association study of bladder cancer identifies a new susceptibility locus within SLC14A1, a urea transporter gene on chromosome 18q12.3. Hum. Mol. Genet. 2011, 20, 4282–4289. [Google Scholar] [CrossRef] [PubMed]
- Koutros, S.; Baris, D.; Fischer, A.; Tang, W.; Garcia-Closas, M.; Karagas, M.R.; Schwenn, M.; Johnson, A.; Figueroa, J.; Waddell, R.; et al. Differential urinary specific gravity as a molecular phenotype of the bladder cancer genetic association in the urea transporter gene, SLC14A1. Int. J. Cancer 2013, 133, 3008–3013. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Funt, S.A.; Rosenberg, J.E. Systemic, perioperative management of muscle-invasive bladder cancer and future horizons. Nat. Rev. Clin. Oncol. 2017, 14, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Soloway, M.S. Bladder cancer: Lack of progress in bladder cancer—What are the obstacles? Nat. Rev. Urol. 2013, 10, 5–6. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e525. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Sjodahl, G.; Jackson, C.L.; Bartlett, J.M.; Siemens, D.R.; Berman, D.M. Molecular profiling in muscle-invasive bladder cancer: More than the sum of its parts. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Faltas, B.M. Treatment resistance in urothelial carcinoma: An evolutionary perspective. Nat. Rev. Clin. Oncol. 2018, 15, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; de Mulder, P.H.; Schornagel, J.H.; Theodore, C.; Fossa, S.D.; van Oosterom, A.T.; Witjes, F.; Spina, M.; van Groeningen, C.J.; de Balincourt, C.; et al. Randomized phase III trial of high-dose-intensity methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) chemotherapy and recombinant human granulocyte colony-stimulating factor versus classic MVAC in advanced urothelial tract tumors: European Organization for Research and Treatment of Cancer Protocol no. 30924. J. Clin. Oncol. 2001, 19, 2638–2646. [Google Scholar] [PubMed]
- von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Apolo, A.B.; Infante, J.R.; Balmanoukian, A.; Patel, M.R.; Wang, D.; Kelly, K.; Mega, A.E.; Britten, C.D.; Ravaud, A.; Mita, A.C.; et al. Avelumab, an Anti-Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. J. Clin. Oncol. 2017, 35, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Callahan, M.K.; Bono, P.; Kim, J.; Spiliopoulou, P.; Calvo, E.; Pillai, R.N.; Ott, P.A.; de Braud, F.; Morse, M.; et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016, 17, 1590–1598. [Google Scholar] [CrossRef]
- Zibelman, M.; Ramamurthy, C.; Plimack, E.R. Emerging role of immunotherapy in urothelial carcinoma-Advanced disease. Urol. Oncol. 2016, 34, 538–547. [Google Scholar] [CrossRef]
- Faltas, B.M.; Prandi, D.; Tagawa, S.T.; Molina, A.M.; Nanus, D.M.; Sternberg, C.; Rosenberg, J.; Mosquera, J.M.; Robinson, B.; Elemento, O.; et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 2016, 48, 1490–1499. [Google Scholar] [CrossRef]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Marmorstein, R. Protein modules that manipulate histone tails for chromatin regulation. Nat. Rev. Mol. Cell Biol. 2001, 2, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- cBioPortal for Cancer Genomics. Available online: http://www.cbioportal.org (accessed on 1 October 2018).
- RCSB PDB Protein Data Bank. Available online: http://www.rcsb.org (accessed on 1 December 2018).
- Ensembl Release 94 (October 2018). Available online: https://www.ensembl.org/Homo_sapiens/Info/Index (accessed on 1 October 2018).
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. Bioessays 2018, 40, e1800078. [Google Scholar] [CrossRef]
- Clements, A.; Rojas, J.R.; Trievel, R.C.; Wang, L.; Berger, S.L.; Marmorstein, R. Crystal structure of the histone acetyltransferase domain of the human PCAF transcriptional regulator bound to coenzyme A. Embo J. 1999, 18, 3521–3532. [Google Scholar] [CrossRef]
- Lin, Y.; Fletcher, C.M.; Zhou, J.; Allis, C.D.; Wagner, G. Solution structure of the catalytic domain of GCN5 histone acetyltransferase bound to coenzyme A. Nature 1999, 400, 86–89. [Google Scholar]
- Delvecchio, M.; Gaucher, J.; Aguilar-Gurrieri, C.; Ortega, E.; Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013, 20, 1040–1046. [Google Scholar] [CrossRef]
- Yan, Y.; Barlev, N.A.; Haley, R.H.; Berger, S.L.; Marmorstein, R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol. Cell 2000, 6, 1195–1205. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. Embo J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Gupta, A.; Guerin-Peyrou, T.G.; Sharma, G.G.; Park, C.; Agarwal, M.; Ganju, R.K.; Pandita, S.; Choi, K.; Sukumar, S.; Pandita, R.K.; et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol. 2008, 28, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Dixon, M.P.; Kueh, A.J.; Voss, A.K. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol. 2008, 28, 5093–5105. [Google Scholar] [CrossRef]
- Chelmicki, T.; Dundar, F.; Turley, M.J.; Khanam, T.; Aktas, T.; Ramirez, F.; Gendrel, A.V.; Wright, P.R.; Videm, P.; Backofen, R.; et al. MOF-associated complexes ensure stem cell identity and Xist repression. Elife 2014, 3, e02024. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Laherty, C.D.; Yang, W.M.; Sun, J.M.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef]
- Zhang, Y.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [PubMed]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Investig. 2000, 105, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. Embo J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. Association of class II histone deacetylases with heterochromatin protein 1: Potential role for histone methylation in control of muscle differentiation. Mol. Cell. Biol. 2002, 22, 7302–7312. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Lu, J.R.; Olson, E.N. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J. Biol. Chem. 2001, 276, 35–39. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; De Francesco, R.; Gallinari, P.; Lahm, A.; Neddermann, P.; Rowley, M.; Serafini, S.; Steinkuhler, C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg. Med. Chem. Lett. 2008, 18, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.D.; Cai, R.; Bhatia, U.; Asselbergs, F.A.; Song, C.; Terry, R.; Trogani, N.; Widmer, R.; Atadja, P.; Cohen, D. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 2002, 277, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Ye, D.; Zhu, S.; Jia, W.; Lu, C.; Wang, G.; Guo, X.; Yu, Y.; Wu, C.; Kang, J. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget 2017, 8, 61338–61349. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. Embo J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef]
- Villagra, A.; Cheng, F.; Wang, H.W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Marin de Evsikova, C.; Bian, K.; Achille, A.; Telles, E.; Pei, H.; Seto, E. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Firestein, R.; Blander, G.; Michan, S.; Oberdoerffer, P.; Ogino, S.; Campbell, J.; Bhimavarapu, A.; Luikenhuis, S.; de Cabo, R.; Fuchs, C.; et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 2008, 3, e2020. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Southwood, C.M.; Peppi, M.; Dryden, S.; Tainsky, M.A.; Gow, A. Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 2007, 32, 187–195. [Google Scholar] [CrossRef]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef]
- Wang, Y.P.; Zhou, L.S.; Zhao, Y.Z.; Wang, S.W.; Chen, L.L.; Liu, L.X.; Ling, Z.Q.; Hu, F.J.; Sun, Y.P.; Zhang, J.Y.; et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. Embo J. 2014, 33, 1304–1320. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell. Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Guarente, L. The SirT3 divining rod points to oxidative stress. Mol. Cell 2011, 42, 561–568. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Karim, M.F.; Sato, Y.; Senokuchi, T.; Miyata, K.; Fukuda, T.; Go, C.; Tasaki, M.; Uchimura, K.; Kadomatsu, T.; et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. 2014, 19, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef]
- Chen, S.; Seiler, J.; Santiago-Reichelt, M.; Felbel, K.; Grummt, I.; Voit, R. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol. Cell 2013, 52, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef]
- Grob, A.; Roussel, P.; Wright, J.E.; McStay, B.; Hernandez-Verdun, D.; Sirri, V. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J. Cell Sci. 2009, 122, 489–498. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 1 October 2018).
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Nagy, L.; Inoue, S.; Shao, W.; Miller, W.H., Jr.; Evans, R.M. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 1998, 391, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Evans, R.M. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell 2000, 5, 821–830. [Google Scholar] [CrossRef]
- Minucci, S.; Maccarana, M.; Cioce, M.; De Luca, P.; Gelmetti, V.; Segalla, S.; Di Croce, L.; Giavara, S.; Matteucci, C.; Gobbi, A.; et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol. Cell 2000, 5, 811–820. [Google Scholar] [CrossRef]
- Licht, J.D. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene 2001, 20, 5660–5679. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheney, M.D.; Gaudet, J.J.; Chruszcz, M.; Lukasik, S.M.; Sugiyama, D.; Lary, J.; Cole, J.; Dauter, Z.; Minor, W.; et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell 2006, 9, 249–260. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 2001, 11, 497–504. [Google Scholar] [CrossRef]
- Zhou, X.; Richon, V.M.; Wang, A.H.; Yang, X.J.; Rifkind, R.A.; Marks, P.A. Histone deacetylase 4 associates with extracellular signal-regulated kinases 1 and 2, and its cellular localization is regulated by oncogenic Ras. Proc. Natl. Acad. Sci. USA 2000, 97, 14329–14333. [Google Scholar] [CrossRef]
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170. [Google Scholar] [CrossRef]
- Masoumi, K.C.; Daams, R.; Sime, W.; Siino, V.; Ke, H.; Levander, F.; Massoumi, R. NLK-mediated phosphorylation of HDAC1 negatively regulates Wnt signaling. Mol. Biol. Cell 2017, 28, 346–355. [Google Scholar] [CrossRef]
- Gonzalez-Zuniga, M.; Contreras, P.S.; Estrada, L.D.; Chamorro, D.; Villagra, A.; Zanlungo, S.; Seto, E.; Alvarez, A.R. c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer’s disease. Mol. Cell 2014, 56, 163–173. [Google Scholar] [CrossRef]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Hanigan, T.W.; Aboukhatwa, S.M.; Taha, T.Y.; Frasor, J.; Petukhov, P.A. Divergent JNK Phosphorylation of HDAC3 in Triple-Negative Breast Cancer Cells Determines HDAC Inhibitor Binding and Selectivity. Cell Chem. Biol. 2017, 24, 1356–1367.e1358. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Choi, J.H.; Kwon, H.J.; Yoon, B.I.; Kim, J.H.; Han, S.U.; Joo, H.J.; Kim, D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Gottlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.; de Paula Queiroz, R.; Valera, E.T.; da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef]
- Saji, S.; Kawakami, M.; Hayashi, S.; Yoshida, N.; Hirose, M.; Horiguchi, S.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar] [CrossRef]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Costa-Machado, L.F.; Martin-Hernandez, R.; Sanchez-Luengo, M.A.; Hess, K.; Vales-Villamarin, C.; Barradas, M.; Lynch, C.; de la Nava, D.; Diaz-Ruiz, A.; de Cabo, R.; et al. Sirt1 protects from K-Ras-driven lung carcinogenesis. Embo Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef]
- Heideman, M.R.; Wilting, R.H.; Yanover, E.; Velds, A.; de Jong, J.; Kerkhoven, R.M.; Jacobs, H.; Wessels, L.F.; Dannenberg, J.H. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013, 121, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Kiefer, F.; Prudenziati, M.; Spiller, C.; Hansen, J.; Floss, T.; Wurst, W.; Minucci, S.; Gottlicher, M. Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 2007, 67, 9047–9054. [Google Scholar] [CrossRef]
- Santoro, F.; Botrugno, O.A.; Dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Pinkerneil, M.; Hoffmann, M.J.; Deenen, R.; Kohrer, K.; Arent, T.; Schulz, W.A.; Niegisch, G. Inhibition of Class I Histone Deacetylases 1 and 2 Promotes Urothelial Carcinoma Cell Death by Various Mechanisms. Mol. Cancer 2016, 15, 299–312. [Google Scholar] [CrossRef]
- Ozawa, A.; Tanji, N.; Kikugawa, T.; Sasaki, T.; Yanagihara, Y.; Miura, N.; Yokoyama, M. Inhibition of bladder tumour growth by histone deacetylase inhibitor. BJU Int. 2010, 105, 1181–1186. [Google Scholar] [CrossRef]
- Pinkerneil, M.; Hoffmann, M.J.; Schulz, W.A.; Niegisch, G. HDACs and HDAC Inhibitors in Urothelial Carcinoma—Perspectives for an Antineoplastic Treatment. Curr. Med. Chem. 2017, 24, 4151–4165. [Google Scholar] [CrossRef] [PubMed]
- Ozdag, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Junqueira-Neto, S.; Vieira, F.Q.; Montezuma, D.; Costa, N.R.; Antunes, L.; Baptista, T.; Oliveira, A.I.; Graca, I.; Rodrigues, A.; Magalhaes, J.S.; et al. Phenotypic impact of deregulated expression of class I histone deacetylases in urothelial cell carcinoma of the bladder. Mol. Carcinog. 2015, 54, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Alivand, M.; Soufi, R.T.; Madani, A.H.; Esmaeili, S.N.; Vaziri, H.R.; Sohani, M.M.; Rafati, M.; Hamami, P.; Ajamian, F. Histonedeacetylase 1 mRNA has elevated expression in clinical specimen of bladder cancer. Bratisl. Lek. Listy 2018, 119, 12–16. [Google Scholar] [CrossRef]
- Zekri, A.R.; Hassan, Z.K.; Bahnassy, A.A.; Khaled, H.M.; El-Rouby, M.N.; Haggag, R.M.; Abu-Taleb, F.M. Differentially expressed genes in metastatic advanced Egyptian bladder cancer. Asian Pac. J. Cancer Prev. 2015, 16, 3543–3549. [Google Scholar] [CrossRef]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef]
- Lehmann, M.; Hoffmann, M.J.; Koch, A.; Ulrich, S.M.; Schulz, W.A.; Niegisch, G. Histone deacetylase 8 is deregulated in urothelial cancer but not a target for efficient treatment. J. Exp. Clin. Cancer Res. 2014, 33, 59. [Google Scholar] [CrossRef]
- Xu, X.S.; Wang, L.; Abrams, J.; Wang, G. Histone deacetylases (HDACs) in XPC gene silencing and bladder cancer. J. Hematol. Oncol. 2011, 4, 17. [Google Scholar] [CrossRef]
- Rosik, L.; Niegisch, G.; Fischer, U.; Jung, M.; Schulz, W.A.; Hoffmann, M.J. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol. 2014, 15, 742–757. [Google Scholar] [CrossRef]
- Thole, T.M.; Lodrini, M.; Fabian, J.; Wuenschel, J.; Pfeil, S.; Hielscher, T.; Kopp-Schneider, A.; Heinicke, U.; Fulda, S.; Witt, O.; et al. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. 2017, 8, e2635. [Google Scholar] [CrossRef] [PubMed]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 132, 2200–2208. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G.; Peng, J.; Qian, G.; Jiang, W.; Xie, C.; Xiao, Y.; Wang, X. Knockdown of SIRT1 Suppresses Bladder Cancer Cell Proliferation and Migration and Induces Cell Cycle Arrest and Antioxidant Response through FOXO3a-Mediated Pathways. Biomed. Res. Int. 2017, 2017, 3781904. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Wu, W.; Li, X.; Zhao, L.; Chen, W. HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol. Rep. 2012, 27, 819–824. [Google Scholar]
- Li, S.; Banck, M.; Mujtaba, S.; Zhou, M.M.; Sugrue, M.M.; Walsh, M.J. p53-induced growth arrest is regulated by the mitochondrial SirT3 deacetylase. PLoS ONE 2010, 5, e10486. [Google Scholar] [CrossRef]
- Wu, M.; Dickinson, S.I.; Wang, X.; Zhang, J. Expression and function of SIRT6 in muscle invasive urothelial carcinoma of the bladder. Int. J. Clin. Exp. Pathol. 2014, 7, 6504–6513. [Google Scholar]
- Han, Y.; Liu, Y.; Zhang, H.; Wang, T.; Diao, R.; Jiang, Z.; Gui, Y.; Cai, Z. Hsa-miR-125b suppresses bladder cancer development by down-regulating oncogene SIRT7 and oncogenic long non-coding RNA MALAT1. Febs Lett. 2013, 587, 3875–3882. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System. Available online: https://pymol.org/2/ (accessed on 14 March 2019).
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef]
- Zhang, Z.; Karam, J.; Frenkel, E.; Sagalowsky, A.; Hsieh, J.T. The application of epigenetic modifiers on the treatment of prostate and bladder cancer. Urol. Oncol. 2006, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Sharma, N.L.; Groselj, B.; Hamdy, F.C.; Kiltie, A.E. The emerging role of histone deacetylase (HDAC) inhibitors in urological cancers. BJU Int. 2013, 111, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef]
- Papeleu, P.; Vanhaecke, T.; Elaut, G.; Vinken, M.; Henkens, T.; Snykers, S.; Rogiers, V. Differential effects of histone deacetylase inhibitors in tumor and normal cells-what is the toxicological relevance? Crit. Rev. Toxicol. 2005, 35, 363–378. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Ruiz-Llorente, L.; Sanchez-Martinez, R.; Garcia, L.; Zambrano, A.; Aranda, A. Histone deacetylase inhibitors: Mechanism of action and therapeutic use in cancer. Clin. Transl. Oncol. 2008, 10, 395–398. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. (Tokyo) 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Karam, J.A.; Fan, J.; Stanfield, J.; Richer, E.; Benaim, E.A.; Frenkel, E.; Antich, P.; Sagalowsky, A.I.; Mason, R.P.; Hsieh, J.T. The use of histone deacetylase inhibitor FK228 and DNA hypomethylation agent 5-azacytidine in human bladder cancer therapy. Int. J. Cancer 2007, 120, 1795–1802. [Google Scholar] [CrossRef]
- Fan, J.; Stanfield, J.; Guo, Y.; Karam, J.A.; Frenkel, E.; Sun, X.; Hsieh, J.T. Effect of trans-2,3-dimethoxycinnamoyl azide on enhancing antitumor activity of romidepsin on human bladder cancer. Clin. Cancer Res. 2008, 14, 1200–1207. [Google Scholar] [CrossRef]
- Holscher, A.S.; Schulz, W.A.; Pinkerneil, M.; Niegisch, G.; Hoffmann, M.J. Combined inhibition of BET proteins and class I HDACs synergistically induces apoptosis in urothelial carcinoma cell lines. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef]
- Li, Q.Q.; Hao, J.J.; Zhang, Z.; Hsu, I.; Liu, Y.; Tao, Z.; Lewi, K.; Metwalli, A.R.; Agarwal, P.K. Histone deacetylase inhibitor-induced cell death in bladder cancer is associated with chromatin modification and modifying protein expression: A proteomic approach. Int. J. Oncol. 2016, 48, 2591–2607. [Google Scholar] [CrossRef]
- Choudhary, S.; Wang, H.C. Proapoptotic ability of oncogenic H-Ras to facilitate apoptosis induced by histone deacetylase inhibitors in human cancer cells. Mol. Cancer 2007, 6, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Wang, H.C. Role of reactive oxygen species in proapoptotic ability of oncogenic H-Ras to increase human bladder cancer cell susceptibility to histone deacetylase inhibitor for caspase induction. J. Cancer Res. Clin. Oncol. 2009, 135, 1601–1613. [Google Scholar] [CrossRef]
- Choudhary, S.; Rathore, K.; Wang, H.C. Differential induction of reactive oxygen species through Erk1/2 and Nox-1 by FK228 for selective apoptosis of oncogenic H-Ras-expressing human urinary bladder cancer J82 cells. J. Cancer Res. Clin. Oncol. 2011, 137, 471–480. [Google Scholar] [CrossRef]
- Philipson, L.; Pettersson, R.F. The coxsackie-adenovirus receptor—A new receptor in the immunoglobulin family involved in cell adhesion. Curr. Top. Microbiol. Immunol. 2004, 273, 87–111. [Google Scholar] [PubMed]
- Sachs, M.D.; Rauen, K.A.; Ramamurthy, M.; Dodson, J.L.; De Marzo, A.M.; Putzi, M.J.; Schoenberg, M.P.; Rodriguez, R. Integrin alpha(v) and coxsackie adenovirus receptor expression in clinical bladder cancer. Urology 2002, 60, 531–536. [Google Scholar] [CrossRef]
- Li, Y.; Pong, R.C.; Bergelson, J.M.; Hall, M.C.; Sagalowsky, A.I.; Tseng, C.P.; Wang, Z.; Hsieh, J.T. Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999, 59, 325–330. [Google Scholar]
- Okegawa, T.; Nutahara, K.; Pong, R.C.; Higashihara, E.; Hsieh, J.T. Enhanced transgene expression in urothelial cancer gene therapy with histone deacetylase inhibitor. J. Urol. 2005, 174, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Sung, J.; Cohen, M.; Chowdhury, W.H.; Sachs, M.D.; Li, Y.; Lakshmanan, Y.; Yung, B.Y.; Lupold, S.E.; Rodriguez, R. Valproic acid inhibits invasiveness in bladder cancer but not in prostate cancer cells. J. Pharm. Exp. 2006, 319, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Vallo, S.; Xi, W.; Hudak, L.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition delays cell cycle progression of human bladder cancer cells in vitro. Anticancer Drugs 2011, 22, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Meyer dos Santos, S.; Schneider, T.; Makarevic, J.; Hudak, L.; Bartsch, G.; Haferkamp, A.; Wiesner, C.; Blaheta, R.A. HDAC inhibition suppresses bladder cancer cell adhesion to collagen under flow conditions. Exp. Biol. Med. (Maywood) 2013, 238, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Byler, T.K.; Leocadio, D.; Shapiro, O.; Bratslavsky, G.; Stodgell, C.J.; Wood, R.W.; Messing, E.M.; Reeder, J.E. Valproic acid decreases urothelial cancer cell proliferation and induces thrombospondin-1 expression. BMC Urol. 2012, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.A.; Wilson, S.S.; Rove, K.O.; Kumar, B.; Koul, S.; Lim, D.D.; Meacham, R.B.; Koul, H.K. Potentiation of mitomycin C tumoricidal activity for transitional cell carcinoma by histone deacetylase inhibitors in vitro. J. Urol. 2011, 186, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jing, Y.; Ouyang, S.; Liu, B.; Zhu, T.; Niu, H.; Tian, Y. Inhibitory effect of valproic acid on bladder cancer in combination with chemotherapeutic agents in vitro and in vivo. Oncol. Lett. 2013, 6, 1492–1498. [Google Scholar] [CrossRef]
- Juengel, E.; Najafi, R.; Rutz, J.; Maxeiner, S.; Makarevic, J.; Roos, F.; Tsaur, I.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition as a treatment concept to combat temsirolimus-resistant bladder cancer cells. Oncotarget 2017, 8, 110016–110028. [Google Scholar] [CrossRef] [PubMed]
- Segura-Pacheco, B.; Avalos, B.; Rangel, E.; Velazquez, D.; Cabrera, G. HDAC inhibitor valproic acid upregulates CAR in vitro and in vivo. Genet. Vaccines 2007, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer 2003, 2, 151–163. [Google Scholar]
- Abedin, S.A.; Thorne, J.L.; Battaglia, S.; Maguire, O.; Hornung, L.B.; Doherty, A.P.; Mills, I.G.; Campbell, M.J. Elevated NCOR1 disrupts a network of dietary-sensing nuclear receptors in bladder cancer cells. Carcinogenesis 2009, 30, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Liu, C.; Pan, J.; Lu, G.; Zhou, Z.; Chen, Z.; Qian, C. Notch3 overexpression enhances progression and chemoresistance of urothelial carcinoma. Oncotarget 2017, 8, 34362–34373. [Google Scholar] [CrossRef] [PubMed]
- Canes, D.; Chiang, G.J.; Billmeyer, B.R.; Austin, C.A.; Kosakowski, M.; Rieger-Christ, K.M.; Libertino, J.A.; Summerhayes, I.C. Histone deacetylase inhibitors upregulate plakoglobin expression in bladder carcinoma cells and display antineoplastic activity in vitro and in vivo. Int. J. Cancer 2005, 113, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; Zhang, X.; Pan, T.J.; Chen, Z.; Ye, Z.Q. Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21WAF1 and G1 cell cycle arrest. Int. J. Urol. 2006, 13, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.F.; Qian, S.B.; Wang, N.; Zhang, L.; Wang, W.M.; Shen, H.B. TRPM2 mediates histone deacetylase inhibition-induced apoptosis in bladder cancer cells. Cancer Biother. Radiopharm. 2015, 30, 87–93. [Google Scholar] [CrossRef]
- Wang, S.C.; Wang, S.T.; Liu, H.T.; Wang, X.Y.; Wu, S.C.; Chen, L.C.; Liu, Y.W. Trichostatin A induces bladder cancer cell death via intrinsic apoptosis at the early phase and Sp1survivin downregulation at the late phase of treatment. Oncol. Rep. 2017, 38, 1587–1596. [Google Scholar] [CrossRef]
- Ou, J.N.; Torrisani, J.; Unterberger, A.; Provencal, N.; Shikimi, K.; Karimi, M.; Ekstrom, T.J.; Szyf, M. Histone deacetylase inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharm. 2007, 73, 1297–1307. [Google Scholar] [CrossRef]
- Yoon, C.Y.; Park, M.J.; Lee, J.S.; Lee, S.C.; Oh, J.J.; Park, H.; Chung, C.W.; Abdullajanov, M.M.; Jeong, S.J.; Hong, S.K.; et al. The histone deacetylase inhibitor trichostatin A synergistically resensitizes a cisplatin resistant human bladder cancer cell line. J. Urol. 2011, 185, 1102–1111. [Google Scholar] [CrossRef]
- Jeon, H.G.; Yoon, C.Y.; Yu, J.H.; Park, M.J.; Lee, J.E.; Jeong, S.J.; Hong, S.K.; Byun, S.S.; Lee, S.E. Induction of caspase mediated apoptosis and down-regulation of nuclear factor-kappaB and Akt signaling are involved in the synergistic antitumor effect of gemcitabine and the histone deacetylase inhibitor trichostatin A in human bladder cancer cells. J. Urol. 2011, 186, 2084–2093. [Google Scholar] [CrossRef]
- Yeh, B.W.; Li, W.M.; Li, C.C.; Kang, W.Y.; Huang, C.N.; Hour, T.C.; Liu, Z.M.; Wu, W.J.; Huang, H.S. Histone deacetylase inhibitor trichostatin A resensitizes gemcitabine resistant urothelial carcinoma cells via suppression of TG-interacting factor. Toxicol. Appl. Pharm. 2016, 290, 98–106. [Google Scholar] [CrossRef]
- Earel, J.K., Jr.; VanOosten, R.L.; Griffith, T.S. Histone deacetylase inhibitors modulate the sensitivity of tumor necrosis factor-related apoptosis-inducing ligand-resistant bladder tumor cells. Cancer Res. 2006, 66, 499–507. [Google Scholar] [CrossRef]
- Sachs, M.D.; Ramamurthy, M.; Poel, H.; Wickham, T.J.; Lamfers, M.; Gerritsen, W.; Chowdhury, W.; Li, Y.; Schoenberg, M.P.; Rodriguez, R. Histone deacetylase inhibitors upregulate expression of the coxsackie adenovirus receptor (CAR) preferentially in bladder cancer cells. Cancer Gene 2004, 11, 477–486. [Google Scholar] [CrossRef] [PubMed]
- El-Zawahry, A.; Lu, P.; White, S.J.; Voelkel-Johnson, C. In vitro efficacy of AdTRAIL gene therapy of bladder cancer is enhanced by trichostatin A-mediated restoration of CAR expression and downregulation of cFLIP and Bcl-XL. Cancer Gene 2006, 13, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Kang, Y.D.; Zhou, M.S.; Fu, L.L.; Hua, Z.H.; Wang, L.M. Experimental study on inhibitory effects of histone deacetylase inhibitor MS-275 and TSA on bladder cancer cells. Urol. Oncol. 2010, 28, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Gould, J.J.; Kenney, P.A.; Rieger-Christ, K.M.; Silva Neto, B.; Wszolek, M.F.; LaVoie, A.; Holway, A.H.; Spurrier, B.; Austin, J.; Cammarata, B.K.; et al. Identification of tumor and invasion suppressor gene modulators in bladder cancer by different classes of histone deacetylase inhibitors using reverse phase protein arrays. J. Urol. 2010, 183, 2395–2402. [Google Scholar] [CrossRef]
- Tang, H.M.; Kuay, K.T.; Koh, P.F.; Asad, M.; Tan, T.Z.; Chung, V.Y.; Lee, S.C.; Thiery, J.P.; Huang, R.J. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016, 2, 16041. [Google Scholar] [CrossRef]
- Lee, S.; Jang, J.; Jeon, H.; Lee, J.; Yoo, S.M.; Park, J.; Lee, M.S. Latent Kaposi’s sarcoma-associated herpesvirus infection in bladder cancer cells promotes drug resistance by reducing reactive oxygen species. J. Microbiol. 2016, 54, 782–788. [Google Scholar] [CrossRef]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Buckley, M.T.; Yoon, J.; Yee, H.; Chiriboga, L.; Liebes, L.; Ara, G.; Qian, X.; Bajorin, D.F.; Sun, T.T.; Wu, X.R.; et al. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J. Transl. Med. 2007, 5, 49. [Google Scholar] [CrossRef]
- Martin, D.T.; Hoimes, C.J.; Kaimakliotis, H.Z.; Cheng, C.J.; Zhang, K.; Liu, J.; Wheeler, M.A.; Kelly, W.K.; Tew, G.N.; Saltzman, W.M.; et al. Nanoparticles for urothelium penetration and delivery of the histone deacetylase inhibitor belinostat for treatment of bladder cancer. Nanomedicine 2013, 9, 1124–1134. [Google Scholar] [CrossRef]
- Molife, L.R.; de Bono, J.S. Belinostat: Clinical applications in solid tumors and lymphoma. Expert Opin. Investig. Drugs 2011, 20, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Groselj, B.; Kerr, M.; Kiltie, A.E. Radiosensitisation of bladder cancer cells by panobinostat is modulated by Ku80 expression. Radiother. Oncol. 2013, 108, 429–433. [Google Scholar] [CrossRef]
- Nicholson, J.; Jevons, S.J.; Groselj, B.; Ellermann, S.; Konietzny, R.; Kerr, M.; Kessler, B.M.; Kiltie, A.E. E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 2017, 77, 3027–3039. [Google Scholar] [CrossRef]
- Ota, S.; Zhou, Z.Q.; Hurlin, P.J. Suppression of FGFR3- and MYC-dependent oncogenesis by tubacin: Association with HDAC6-dependent and independent activities. Oncotarget 2018, 9, 3172–3187. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- NIH U.S. National Library of Medicine ClinicalTrials.gov Database. Available online: https://clinicaltrials.gov (accessed on 1 January 2019).
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer 2013, 12, 2078–2087. [Google Scholar] [CrossRef]
- Eot-Houllier, G.; Fulcrand, G.; Magnaghi-Jaulin, L.; Jaulin, C. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009, 274, 169–176. [Google Scholar] [CrossRef]
- Munster, P.; Marchion, D.; Bicaku, E.; Lacevic, M.; Kim, J.; Centeno, B.; Daud, A.; Neuger, A.; Minton, S.; Sullivan, D. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: Phase I/II trial of valproic acid and epirubicin/FEC. Clin. Cancer Res. 2009, 15, 2488–2496. [Google Scholar] [CrossRef]
- Vitfell-Rasmussen, J.; Judson, I.; Safwat, A.; Jones, R.L.; Rossen, P.B.; Lind-Hansen, M.; Knoblauch, P.; Krarup-Hansen, A. A Phase I/II Clinical Trial of Belinostat (PXD101) in Combination with Doxorubicin in Patients with Soft Tissue Sarcomas. Sarcoma 2016, 2016, 2090271. [Google Scholar] [CrossRef]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef]
- Karagiannis, T.C.; El-Osta, A. Modulation of cellular radiation responses by histone deacetylase inhibitors. Oncogene 2006, 25, 3885–3893. [Google Scholar] [CrossRef]
- Decker, R.H.; Gettinger, S.N.; Glazer, P.M.; Wilson, L.D. Vorinostat, a Histone Deacetylase Inhibitor, in Combination with Thoracic Radiotherapy in Advanced Non-small Cell Lung Cancer: A Dose Escalation Study. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, S574–S575. [Google Scholar] [CrossRef]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef]
- Lassen, U.; Molife, L.R.; Sorensen, M.; Engelholm, S.A.; Vidal, L.; Sinha, R.; Penson, R.T.; Buhl-Jensen, P.; Crowley, E.; Tjornelund, J.; et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br. J. Cancer 2010, 103, 12–17. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Parise, R.A.; Ramanathan, R.K.; Lagattuta, T.F.; Musguire, L.A.; Stoller, R.G.; Potter, D.M.; Argiris, A.E.; Zwiebel, J.A.; Egorin, M.J.; et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin. Cancer Res. 2007, 13, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenet. 2017, 9, 59. [Google Scholar] [CrossRef]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Lee, M.J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The interplay of epigenetic therapy and immunity in locally recurrent or metastatic estrogen receptor-positive breast cancer: Correlative analysis of ENCORE 301, a randomized, placebo-controlled phase II trial of exemestane with or without entinostat. Oncoimmunology 2016, 5, e1219008. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Quinn, D.I.; Hammers, H.J.; Monk, P.; George, S.; Dorff, T.B.; Olencki, T.; Shen, L.; Orillion, A.; Lamonica, D.; et al. Immunomodulation by Entinostat in Renal Cell Carcinoma Patients Receiving High-Dose Interleukin 2: A Multicenter, Single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin. Cancer Res. 2017, 23, 7199–7208. [Google Scholar]
- Grivas, P.; Mortazavi, A.; Picus, J.; Hahn, N.M.; Milowsky, M.I.; Hart, L.L.; Alva, A.; Bellmunt, J.; Pal, S.K.; Bambury, R.M.; et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer 2018. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Cancer Genome Atlas (TCGA)—Bladder Cancer | |||
|---|---|---|---|
| Family Member | Protein Change | Mutation Type | Somatic Mutation Frequency |
| HDAC1 | Q364*, M64I, L276I, Y204S | N, M, M, M | 1.0% |
| HDAC2 | E204K, H180R, R446K | M, M, M | 0.7% |
| HDAC3 | F8L | M | 0.2% |
| HDAC8 | E23K, R361Q, V175M | M, M, M | 0.7% |
| HDAC4 | E587Q, D54N, S290F | M, M, M | 0.7% |
| HDAC5 | Q468*, Q88L, E620K, E591Q, E614K, S886C | N, M, M, M, M, M | 1.2% |
| HDAC7 | D937N, G36Afs*11, M828I, P320Rfs*3 | M, FS del, M, FS del | 1.0% |
| HDAC9 | H935L, Q710H, E49*, S1038F, S3T, G1022Afs*22, G133V, L411V, S522F | M, M, N, M, M, FS del, M, M, M | 2.2% |
| HDAC6 | R236L, H1115R, G344E, G1014W, E333Q, E432Q, E562D, W705*, D1128H, R480C, T260A, V874F | M, M, M, M, M, M, M, N, M, M, M, M | 2.2% |
| HDAC10 | S545C, A434V, R411H | M, M, M | 0.7% |
| HDAC11 | R206H, N47H, D282H, V19M, H35Q | M, M, M, M, M | 1.2% |
| SIRT1 | H471Y, A139del, Y650D, Q632H, E230K, D197H, S550*, E609A, G618E, E164K | M, IF del, M, M, M, M, N, M, M, M | 2.4% |
| SIRT2 | R346Q, R163C | M, M | 0.5% |
| SIRT3 | H217R, G161R | M, M | 0.5% |
| SIRT4 | S208*, L227V, S302C | N, M, M | 0.2% |
| SIRT5 | R44Q, L246V, E125K | M, M, M | 0.7% |
| SIRT6 | E139K, E131Q | M, M | 0.5% |
| SIRT7 | A258Cfs*31, E203K, E203K, R289W | FS ins, M, M, M | 1.0% |
| HDACi Agent | Combined Agent | Phase | Status | Patients | Outcome | Identifier |
|---|---|---|---|---|---|---|
| Vorinostat | - | Phase-II | Terminated | Locally recurrent, or metastatic cancer of the urothelium | Limited clinical benefit; Serious adverse effects (43%) | NCT00363883 |
| Mocetinostat | - | Phase-II | Completed | BlCa patients with inactivating mutations or deletions in the CREBBP and/or EP300 acetyltransferase genes | Severe toxicities; Limited clinical efficacy | NCT02236195 |
| Belinostat | Carboplatin or paclitaxel | Phase-I/II | Completed | BlCa patients | Complete or partial responses in 4 out of 15 BlCa patients enrolled | NCT00421889 |
| Vorinostat | Pembrolizumab | Phase-I/Ib | Ongoing | Advanced urothelial cell carcinoma | NCT02619253 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulou, A.F.; Velentzas, A.D.; Konstantakou, E.G.; Avgeris, M.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Mpakou, V.E.; Iconomidou, V.; Anastasiadou, E.; et al. Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. Int. J. Mol. Sci. 2019, 20, 1291. https://doi.org/10.3390/ijms20061291
Giannopoulou AF, Velentzas AD, Konstantakou EG, Avgeris M, Katarachia SA, Papandreou NC, Kalavros NI, Mpakou VE, Iconomidou V, Anastasiadou E, et al. Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. International Journal of Molecular Sciences. 2019; 20(6):1291. https://doi.org/10.3390/ijms20061291
Chicago/Turabian StyleGiannopoulou, Aikaterini F., Athanassios D. Velentzas, Eumorphia G. Konstantakou, Margaritis Avgeris, Stamatia A. Katarachia, Nikos C. Papandreou, Nikolas I. Kalavros, Vassiliki E. Mpakou, Vassiliki Iconomidou, Ema Anastasiadou, and et al. 2019. "Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer" International Journal of Molecular Sciences 20, no. 6: 1291. https://doi.org/10.3390/ijms20061291
APA StyleGiannopoulou, A. F., Velentzas, A. D., Konstantakou, E. G., Avgeris, M., Katarachia, S. A., Papandreou, N. C., Kalavros, N. I., Mpakou, V. E., Iconomidou, V., Anastasiadou, E., Kostakis, I. K., Papassideri, I. S., Voutsinas, G. E., Scorilas, A., & Stravopodis, D. J. (2019). Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. International Journal of Molecular Sciences, 20(6), 1291. https://doi.org/10.3390/ijms20061291