Integrated Analysis of miRNA and mRNA Expression Profiles in Spleen of Specific Pathogen-Free Chicken Infected with Avian Reticuloendotheliosis Virus Strain SNV


Abstract
:1. Introduction
2. Results
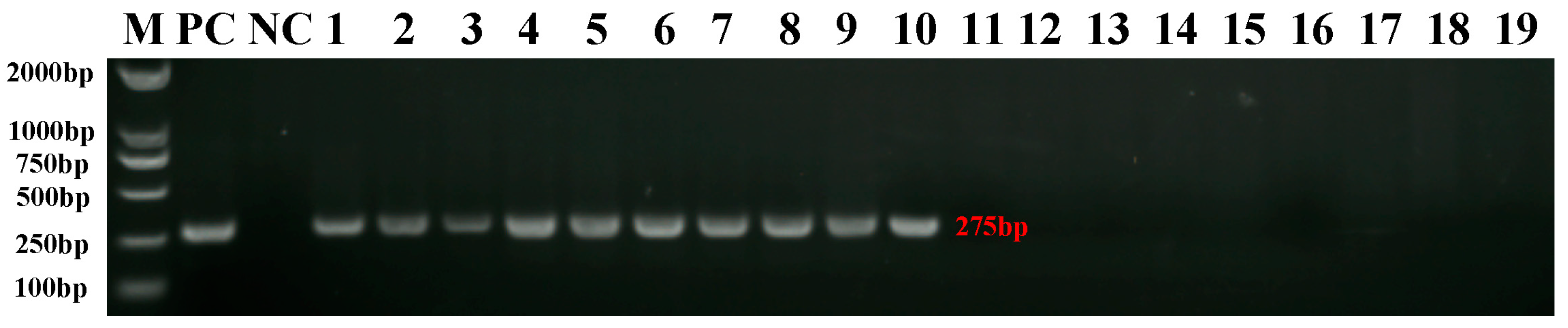
2.1. Verification of REV Infection in SPF Chickens
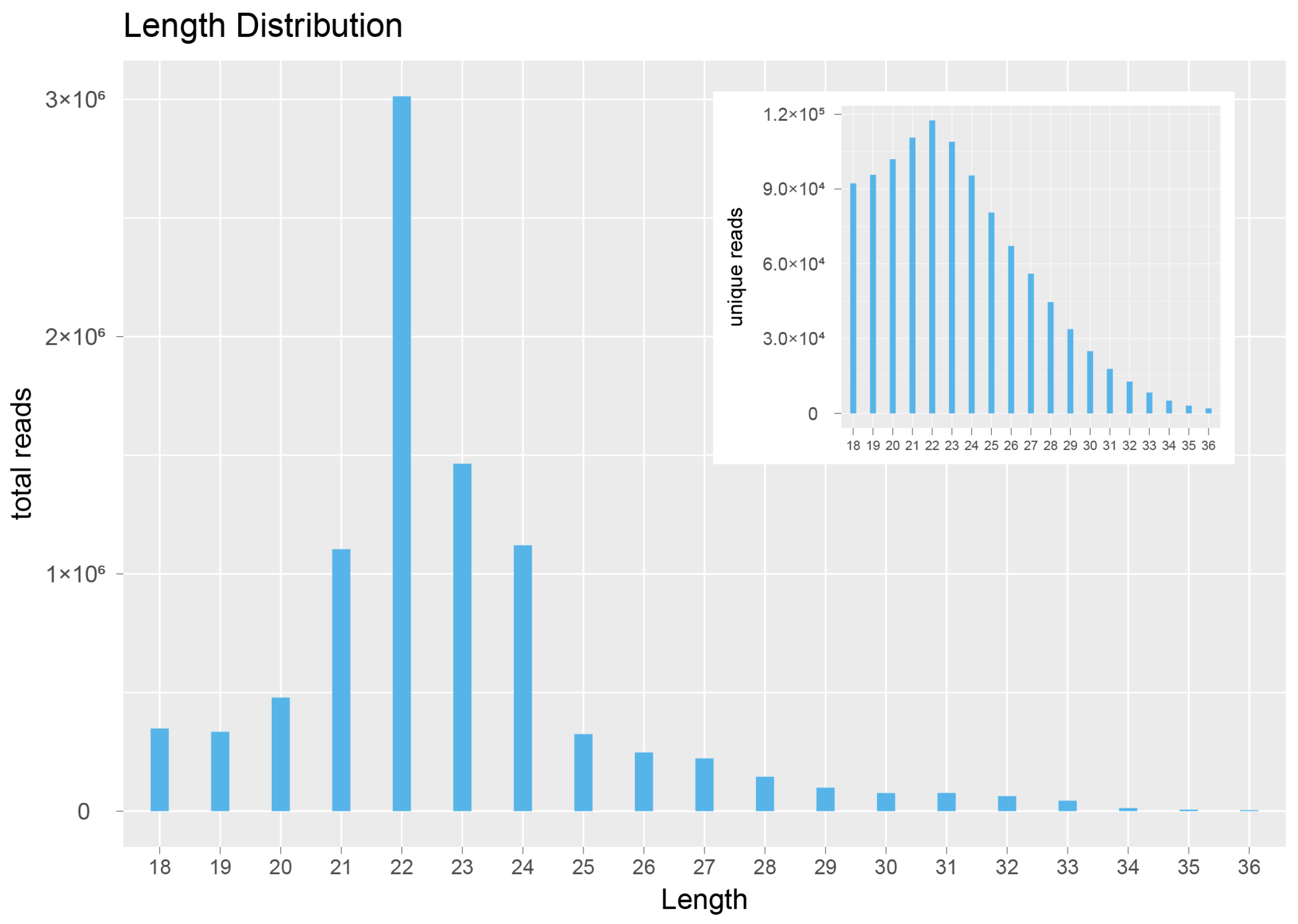
2.2. Analysis of NGS Data from 19 sRNA Libraries
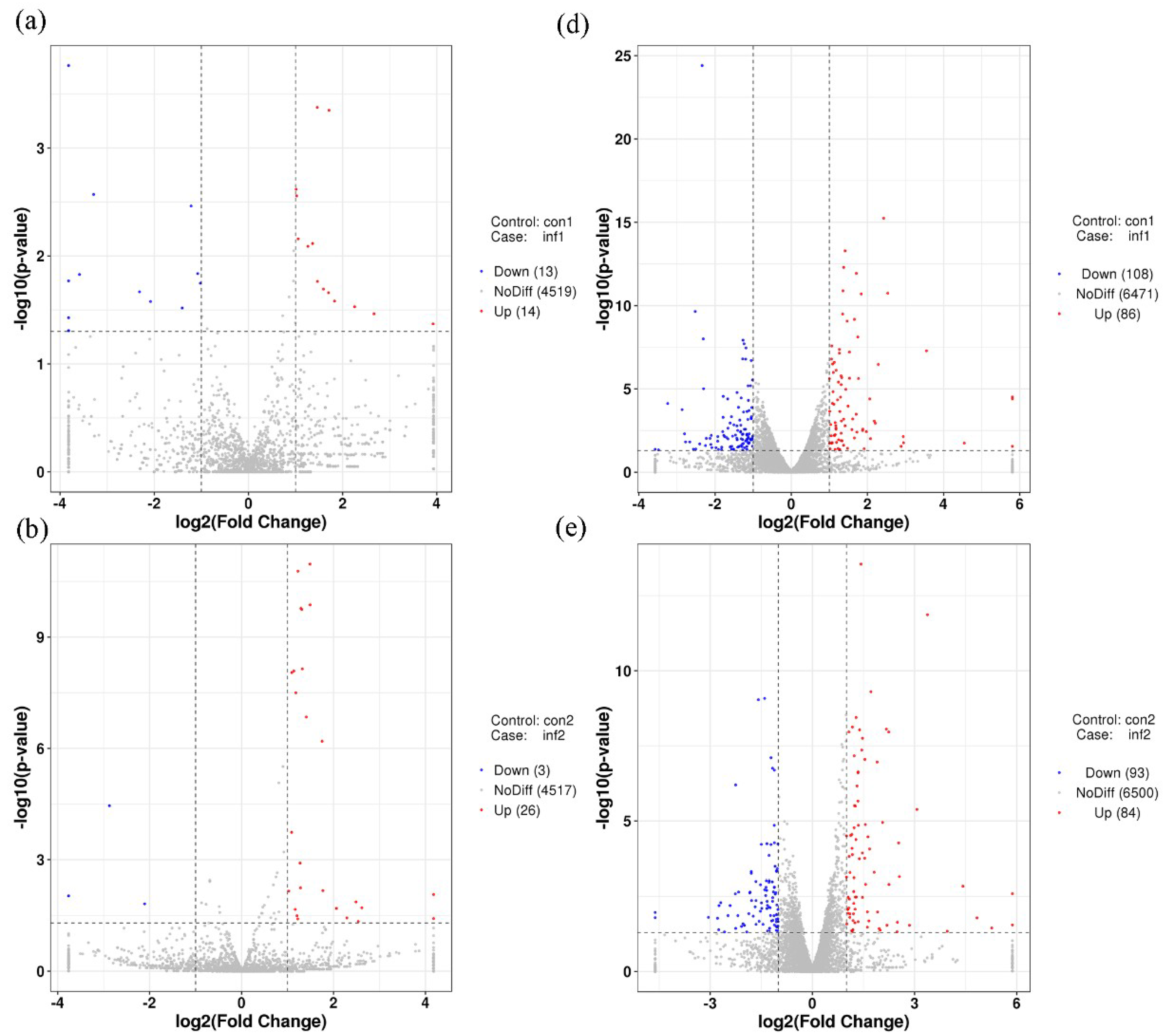
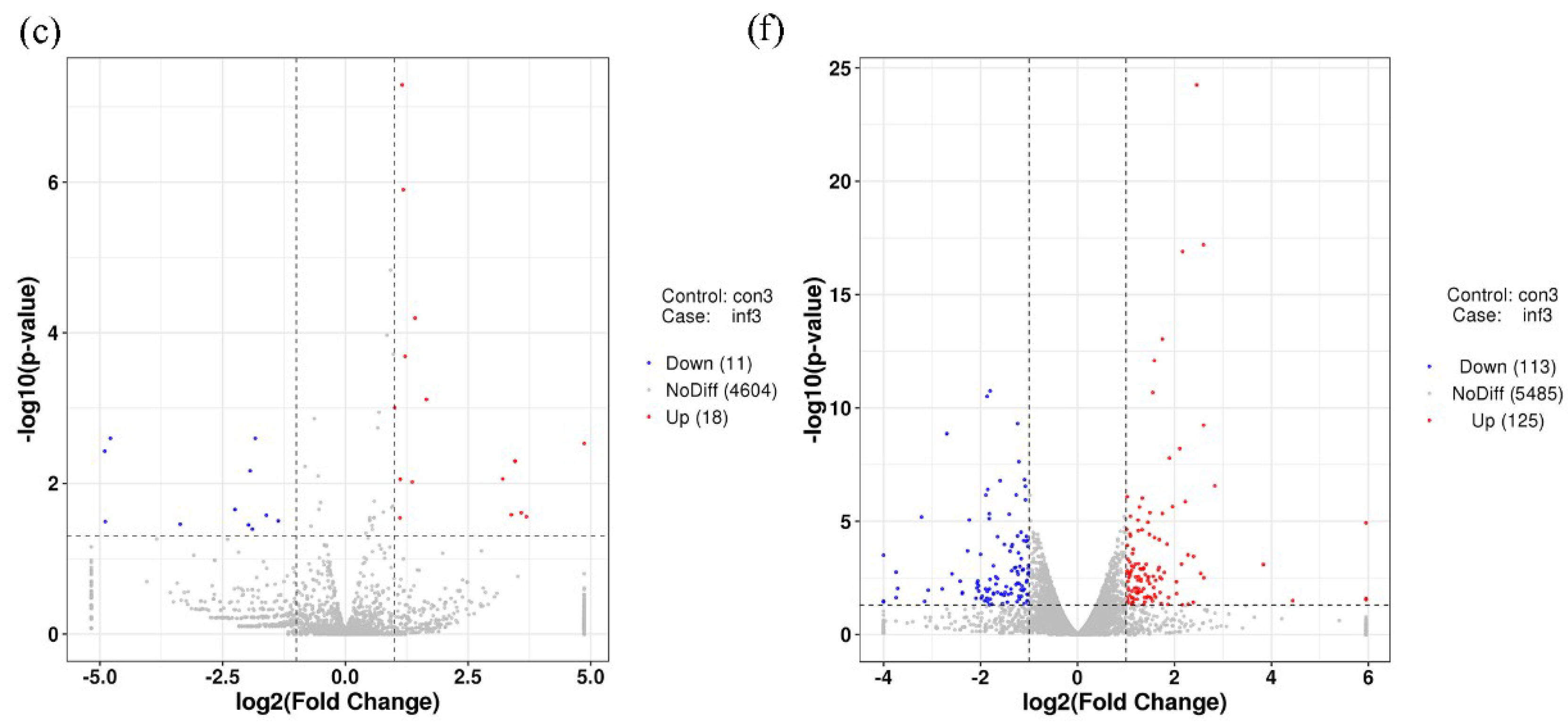
2.3. Gene Expression Analysis and Target Prediction
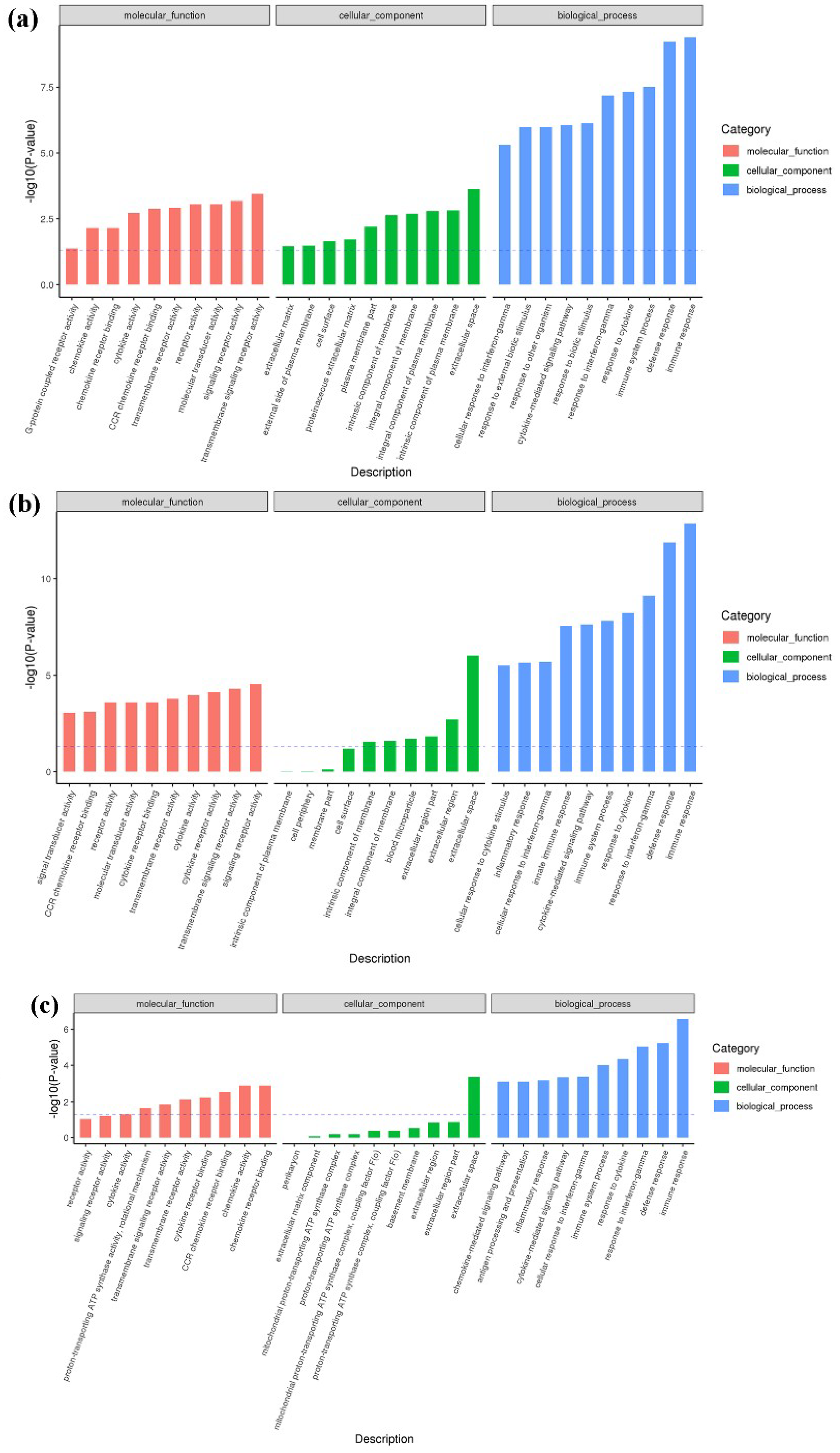
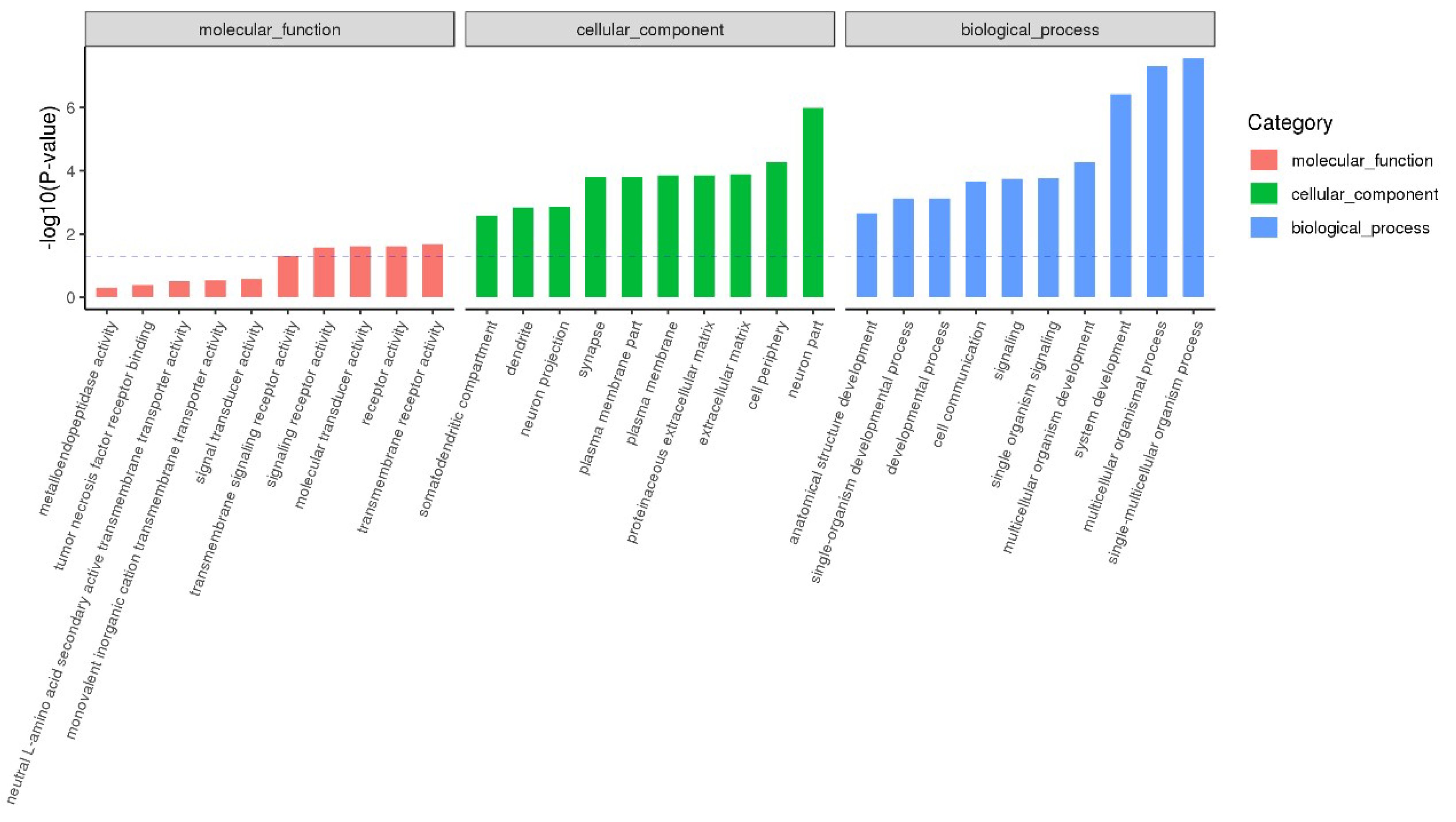
2.4. Functional Enrichment Analysis of DEmiRNA
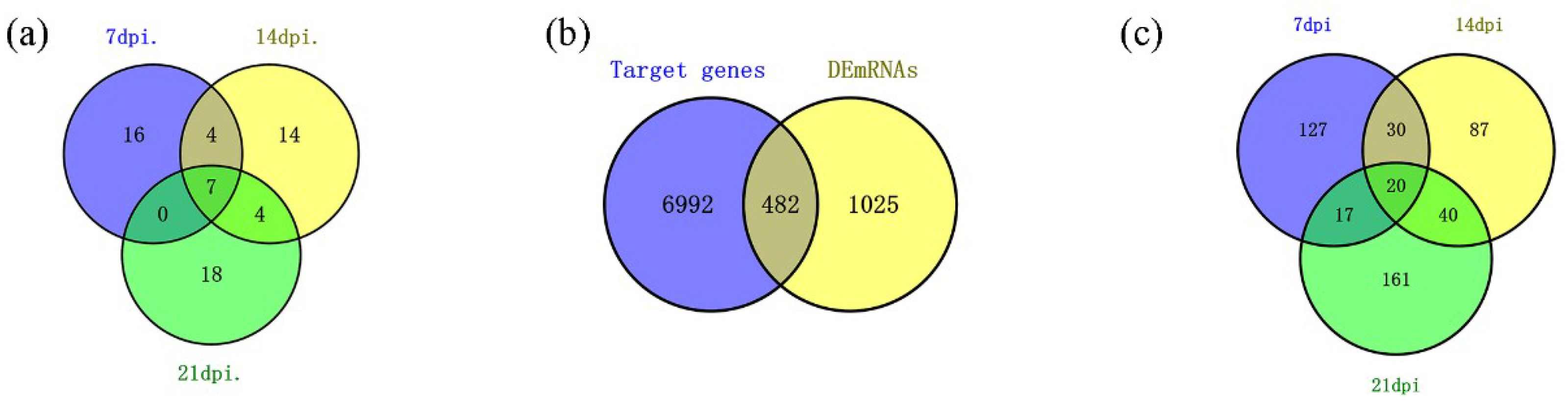
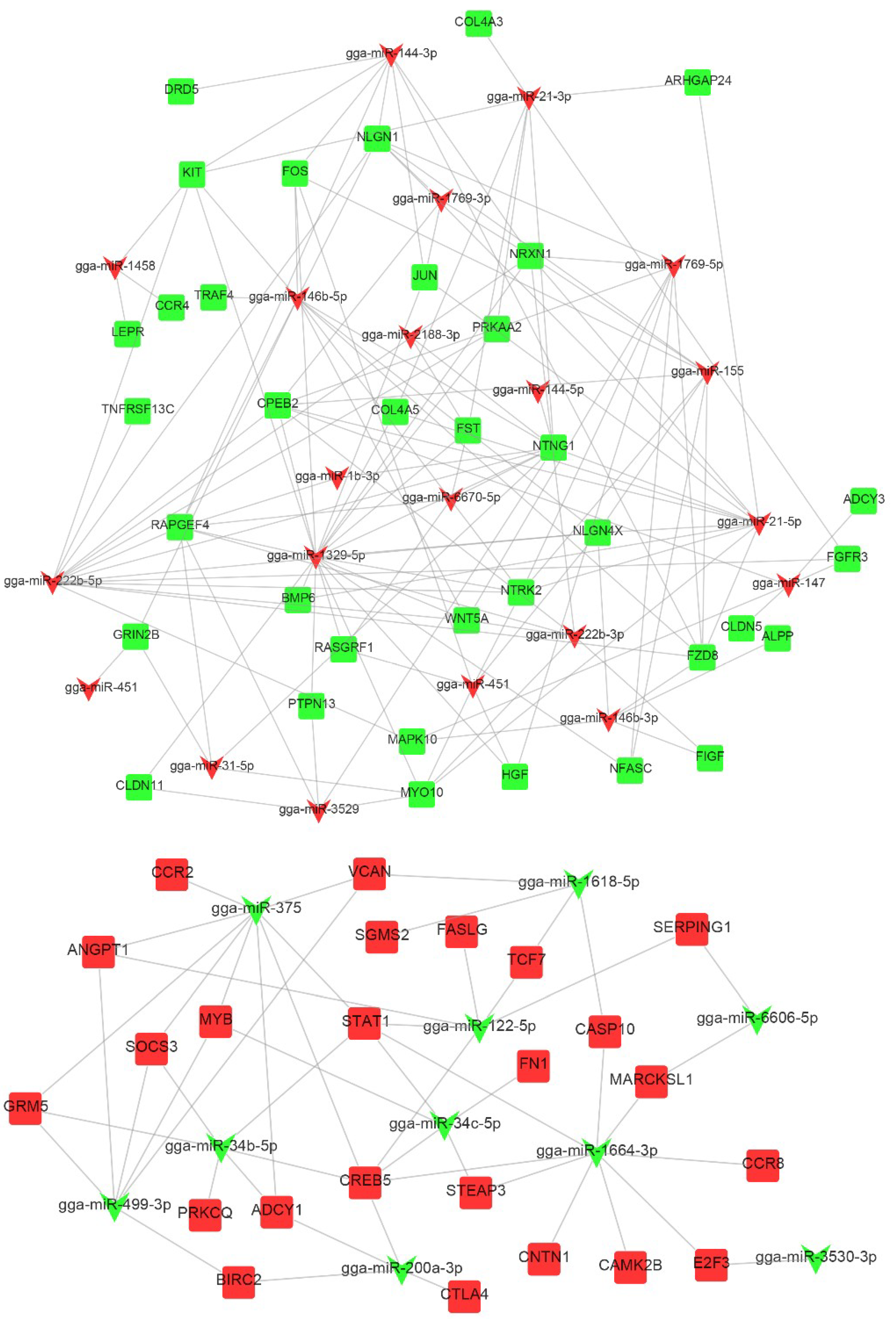
2.5. miRNA–mRNA Integrated Analysis
2.6. Validation of DEmiRNAs and Targets by Quantitative RT-PCR (qRT-PCR)
3. Discussion
4. Materials and Methods
4.1. Ethics Statements
4.2. REV Infections and Sample Collection
4.3. Total RNA Isolation and Virus Infection Assays
4.4. Library Construction and NGS
4.5. Analysis of sRNA Sequencing Data
4.6. Integrative Analyses of DEmiRNAs and DEmRNAs
4.7. The Expression of miRNAs and Their Targets was Validated by qRT-PCR
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bi, Y.; Xu, L.; Qiu, L.; Wang, S.; Liu, X.; Zhang, Y.; Chen, Y.; Zhang, Y.; Xu, Q.; Chang, G.; et al. Reticuloendotheliosis Virus Inhibits the Immune Response Acting on Lymphocytes from Peripheral Blood of Chicken. Front. Physiol. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, A.; Abdul-Cader, M.S.; Almatrouk, Z.; van der Meer, F.; Cork, S.C.; Gomis, S.; Abdul-Careem, M.F. Induction of innate host responses characterized by production of interleukin (IL)-1beta and recruitment of macrophages to the respiratory tract of chickens following infection with infectious bronchitis virus (IBV). Vet. Microbiol. 2018, 215, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, F.R.; Twiehaus, M.J. Isolation of tha avian reticuloendothelial virus (strain T). Avian Dis. 1974, 18, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-J.; Li, H.-M.; Cheng, Z.-Q.; Liu, J.-Z.; Cui, Z.-Z. Influence of REV and ALV-J Co-Infection on Immunologic Function of T Lymphocytes and Histopathology in Broiler Chickens. Agric. Sci. China 2010, 9, 1667–1676. [Google Scholar] [CrossRef]
- Niewiadomska, A.M.; Gifford, R.J. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Shi, Y.; Zhang, L.; Zhu, G.; Diao, X.; Cui, Z. Occurrence of reticuloendotheliosis in Chinese partridge. J. Vet. Med. Sci. 2007, 69, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Zhao, Y.; Hu, S.; Shi, X.; Cui, H.; Wang, Y. Analysis of the spleen proteome of chickens infected with reticuloendotheliosis virus. Arch. Virol. 2017, 162, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, G.; Chiantore, M.V.; Mangino, G.; Romeo, G. MicroRNAs in virus-induced tumorigenesis and IFN system. Cytokine Growth Factor Rev. 2015, 26, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Raisch, J.; Darfeuille-Michaud, A.; Nguyen, H.T. Role of microRNAs in the immune system, inflammation and cancer. World J. Gastroenterol. 2013, 19, 2985–2996. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Trager, W. A new virus of ducks interfering with development of malaria parasite (Plasmodium lophurae). Proc. Soc. Exp. Biol. Med. 1959, 101, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H.; Lee, S.W. The role of microRNAs in hepatitis C virus replication and related liver diseases. J. Microbiol. 2014, 52, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Luo, J.; Zhang, H.; Chang, S.; Song, J. MiRNA expression signatures induced by Marek’s disease virus infection in chickens. Genomics 2012, 99, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Li, X.; Zhao, C.; Han, B.; Qu, L.; Song, J.; Liu, C.; Yang, N. Chicken gga-miR-181a targets MYBL1 and shows an inhibitory effect on proliferation of Marek’s disease virus-transformed lymphoid cell line. Poult. Sci. 2015, 94, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Qu, L.; Chen, Y.; Lamont, S.J.; Yang, N. A systematic analysis of miRNA transcriptome in Marek’s disease virus-induced lymphoma reveals novel and differentially expressed miRNAs. PLoS ONE 2012, 7, e51003. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Zhang, D.; Wang, Q.; Yang, N.; Qu, L. The inhibitory effects of gga-miR-199-3p, gga-miR-140-3p, and gga-miR-221-5p in Marek’s disease tumorigenesis. Poult. Sci. 2015, 94, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ji, J.; Xie, Q.; Shang, H.; Zhang, H.; Xin, X.; Chen, F.; Sun, B.; Xue, C.; Ma, J.; et al. Aberrant expression of liver microRNA in chickens infected with subgroup J avian leukosis virus. Virus Res. 2012, 169, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, Y.; Ji, X.; Qi, X.; Qin, L.; Gao, H.; Wang, Y.; Wang, X. Differential expression of microRNAs in avian leukosis virus subgroup J-induced tumors. Vet. Microbiol. 2013, 162, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ji, J.; Yan, Y.; Lin, W.; Li, H.; Chen, F.; Liu, Y.; Chen, W.; Bi, Y.; Xie, Q. Role of gga-miR-221 and gga-miR-222 during Tumour Formation in Chickens Infected by Subgroup J Avian Leukosis Virus. Viruses 2015, 7, 6538–6551. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shang, H.; Shu, D.; Zhang, H.; Ji, J.; Sun, B.; Li, H.; Xie, Q. gga-miR-375 plays a key role in tumorigenesis post subgroup J avian leukosis virus infection. PLoS ONE 2014, 9, e90878. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Zhang, Y.P.; Li, Y.; Zheng, H.W.; Zheng, Y.S.; Liu, C.J. Distinct expression pattern of miRNAs in Marek’s disease virus infected-chicken splenic tumors and non-tumorous spleen tissues. Res. Vet. Sci. 2014, 97, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.-Y.; Temin, H.M. Lack of Sequence Homology Among RNAs of Avian Leukosis-Sarcoma Viruses, Reticuloendotheliosis Viruses, and Chicken Endogenous RNA-Directed DNA Polymerase Activity. J. Virol. 1973, 12, 1314. [Google Scholar] [PubMed]
- Temin, H.M.; Kassner, V.K. Replication of reticuloendotheliosis viruses in cell culture: acute infection. J. Virol. 1974, 13, 291–297. [Google Scholar] [PubMed]
- Barbacid, M.; Hunter, E.; Aaronson, S.A. Avian reticuloendotheliosis viruses: Evolutionary linkage with mammalian type C retroviruses. J. Virol. 1979, 30, 508–514. [Google Scholar] [PubMed]
- Watanabe, S.; Temin, H.M. Encapsidation sequences for spleen necrosis virus, an avian retrovirus, are between the 5’ long terminal repeat and the start of the gag gene. Proc. Natl. Acad. Sci. USA 1982, 79, 5986. [Google Scholar] [CrossRef] [PubMed]
- Witter, R.L.; Crittenden, L.B. Lymphomas resembling lymphoid leukosis in chickens inoculated with reticuloendotheliosis virus. Int. J. Cancer 1979, 23, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiao, H.; Zhao, J.; Wang, X.; Sun, S.; Lin, H. Allicin Alleviates Reticuloendotheliosis Virus-Induced Immunosuppression via ERK/Mitogen-Activated Protein Kinase Pathway in Specific Pathogen-Free Chickens. Front. Immunol. 2017, 8, 1856. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Bao, Y.; Ye, J.; Shao, H.; Qian, K.; Qin, A. Transcriptional Profiling of Host Gene Expression in Chicken Embryo Fibroblasts Infected with Reticuloendotheliosis Virus Strain HA1101. PLoS ONE 2015, 10, e0126992. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Gao, X.; Liu, C.; Lv, X.; Zheng, S. Analysis of microRNA expression profile in specific pathogen-free chickens in response to reticuloendotheliosis virus infection. Appl. Microbiol. Biotechnol. 2017, 101, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Gao, C.; Fu, L.; Jing, L.; Dang, S.; Zheng, S. Integrative Analyses of Transcriptome Sequencing Identify Functional miRNAs in the Chicken Embryo Fibroblasts Cells Infected with Reticuloendotheliosis Virus. Front. Genet. 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Xue, J.; He, S.; Du, X.; Zhou, J.; Li, C.; Huang, L.; Nair, V.; Yao, Y.; Cheng, Z. Reticuloendotheliosis virus and avian leukosis virus subgroup J synergistically increase the accumulation of exosomal miRNAs. Retrovirology 2018, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A.J.I. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Losi, C.G.; Silini, A.; Fiorini, C.; Soresina, A.; Meini, A.; Ferrari, S.; Notarangelo, L.D.; Lougaris, V.; Plebani, A. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J. Clin. Immunol. 2005, 25, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, D.A.; Gregory, B.; Hoffman, B. AP-1 (Fos/Jun) transcription factors in hematopoietic differentiation and apoptosis. Int. J. Oncol. 1998, 12, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Castellano, R.; Vire, B.; Pion, M.; Quivy, V.; Olive, D.; Hirsch, I.; Van Lint, C.; Collette, Y. Active transcription of the human FASL/CD95L/TNFSF6 promoter region in T lymphocytes involves chromatin remodeling: Role of DNA methylation and protein acetylation suggest distinct mechanisms of transcriptional repression. J. Biol. Chem. 2006, 281, 14719–14728. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Grandoch, M.; Roscioni, S.S.; Schmidt, M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br. J. Pharmacol. 2010, 159, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Abdelli, S.; Puyal, J.; Bielmann, C.; Buchillier, V.; Abderrahmani, A.; Clarke, P.G.H.; Beckmann, J.S.; Bonny, C.J.D. JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia 2009, 52, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Li, H.; Cui, Y.; Wong, A.H.Y.; Langford, C.; Tao, Q. Epigenetic disruption of two proapoptotic genes MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas through hypermethylation of a common bidirectional promoter. Leukemia 2006, 20, 1173. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, S.D. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinform. 2011, 12. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Z. Trusted channels with password-based authentication and TPM-based attestation. In Proceedings of the 2010 International Conference on Communications and Mobile Computing, Caen, France, 28 June–2 July 2010; pp. 223–227. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Read | Clean Reads (≥18 nt) | Mature miRNAs | miRNA Precusors | Q20(%) | Q30(%) |
|---|---|---|---|---|---|---|
| con1-1 | 21,210,076 | 16,264,810 | 476 | 374 | 95.46 | 92.91 |
| con1-2 | 32,179,715 | 18,018,370 | 484 | 384 | 95.46 | 92.92 |
| con1-3 | 11,032,471 | 7,661,330 | 401 | 313 | 95.48 | 92.03 |
| inf1-1 | 10,502,004 | 9,185,246 | 428 | 343 | 95.07 | 93.05 |
| inf1-2 | 10,628,615 | 7,875,010 | 389 | 315 | 95.37 | 92.62 |
| inf1-3 | 22,783,772 | 15,997,877 | 486 | 389 | 95.57 | 92.01 |
| con2-1 | 17,989,084 | 14138332 | 444 | 350 | 96.66 | 92.34 |
| con2-2 | 14,349,826 | 11,647,721 | 416 | 338 | 96.70 | 92.37 |
| con2-3 | 12,940,512 | 10,611,534 | 408 | 324 | 96.39 | 92.81 |
| inf2-1 | 16,402,412 | 13,810,002 | 429 | 340 | 95.03 | 93.98 |
| inf2-2 | 16,231,649 | 13,895,082 | 434 | 347 | 94.10 | 92.29 |
| inf2-3 | 16,781,428 | 14,092,339 | 433 | 339 | 94.36 | 92.65 |
| con3-1 | 12,330,618 | 10,047,514 | 413 | 325 | 95.74 | 92.34 |
| con3-2 | 11,178,524 | 9,091,076 | 403 | 315 | 95.82 | 92.53 |
| con3-3 | 9,816,714 | 8,692,449 | 425 | 336 | 95.55 | 93.25 |
| inf3-1 | 16,059,318 | 13,727,564 | 438 | 350 | 96.43 | 92.85 |
| inf3-2 | 15,587,419 | 13,357,222 | 430 | 341 | 96.54 | 92.13 |
| inf3-3 | 15,102,237 | 12,748,493 | 434 | 343 | 96.57 | 92.17 |
| inf3-4 | 10,884,563 | 9,182,693 | 397 | 317 | 96.72 | 92.44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Jiang, H.; Sun, J.; Diao, Y.; Tang, Y.; Hu, J. Integrated Analysis of miRNA and mRNA Expression Profiles in Spleen of Specific Pathogen-Free Chicken Infected with Avian Reticuloendotheliosis Virus Strain SNV. Int. J. Mol. Sci. 2019, 20, 1041. https://doi.org/10.3390/ijms20051041
Gao S, Jiang H, Sun J, Diao Y, Tang Y, Hu J. Integrated Analysis of miRNA and mRNA Expression Profiles in Spleen of Specific Pathogen-Free Chicken Infected with Avian Reticuloendotheliosis Virus Strain SNV. International Journal of Molecular Sciences. 2019; 20(5):1041. https://doi.org/10.3390/ijms20051041
Chicago/Turabian StyleGao, Shuo, Hao Jiang, Jie Sun, Youxiang Diao, Yi Tang, and Jingdong Hu. 2019. "Integrated Analysis of miRNA and mRNA Expression Profiles in Spleen of Specific Pathogen-Free Chicken Infected with Avian Reticuloendotheliosis Virus Strain SNV" International Journal of Molecular Sciences 20, no. 5: 1041. https://doi.org/10.3390/ijms20051041
APA StyleGao, S., Jiang, H., Sun, J., Diao, Y., Tang, Y., & Hu, J. (2019). Integrated Analysis of miRNA and mRNA Expression Profiles in Spleen of Specific Pathogen-Free Chicken Infected with Avian Reticuloendotheliosis Virus Strain SNV. International Journal of Molecular Sciences, 20(5), 1041. https://doi.org/10.3390/ijms20051041