Characterizing Cellular Responses During Oncolytic Maraba Virus Infection

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
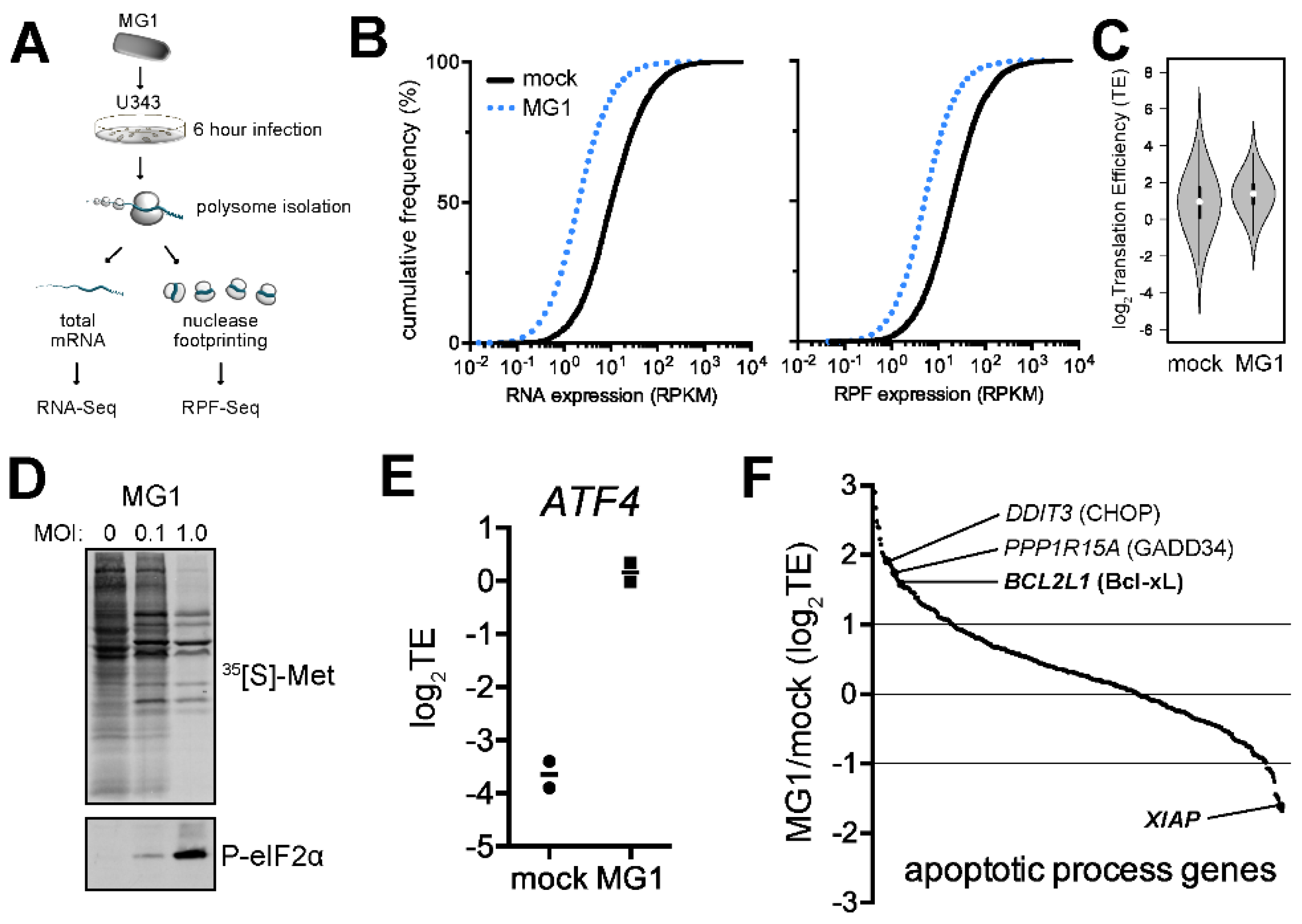
2.1. Ribosome Profiling of MG1-Infected U343 Cells Highlights Cellular Targets of eIF2α Phosphorylation and Apoptosis
2.2. MG1 Regulates Selective Translation of Viral mRNA and Bcl-xL Upon the Inhibition of Global Translation
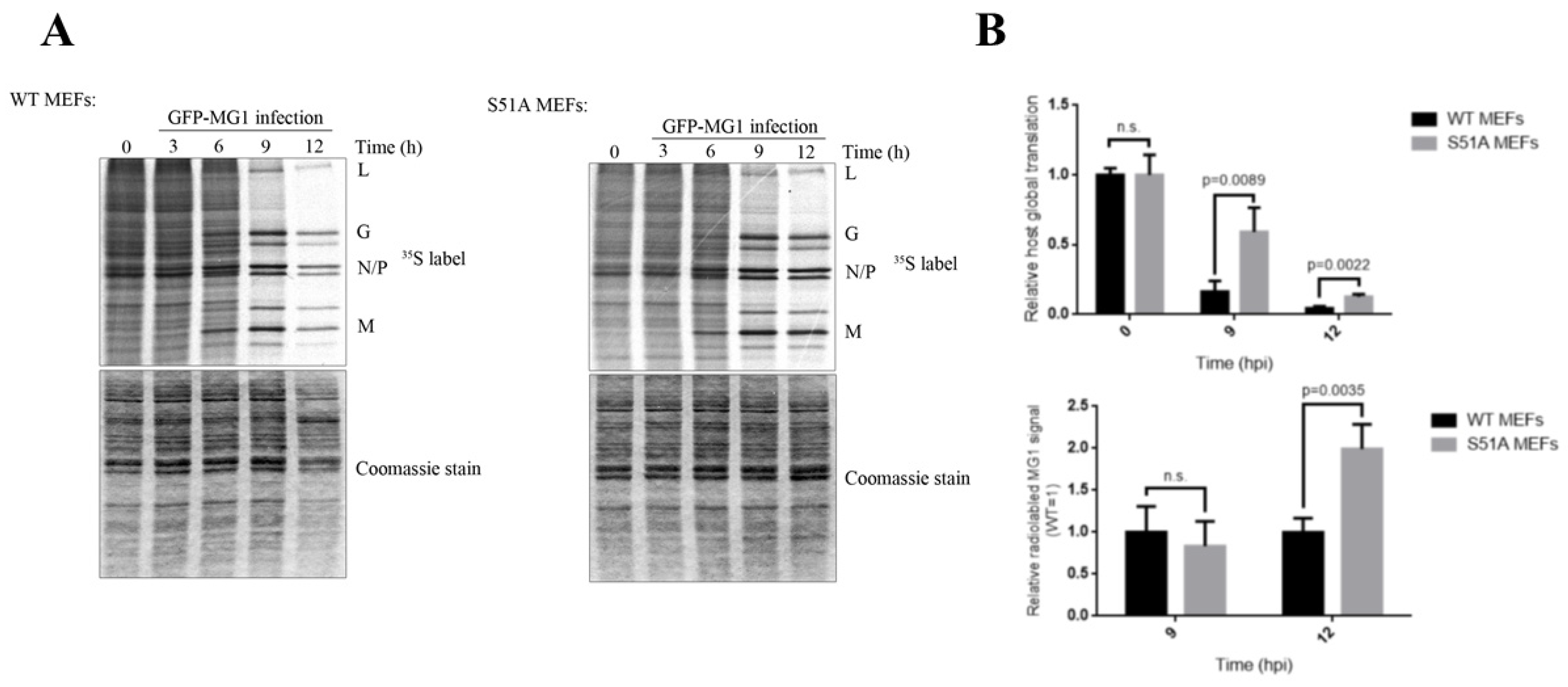
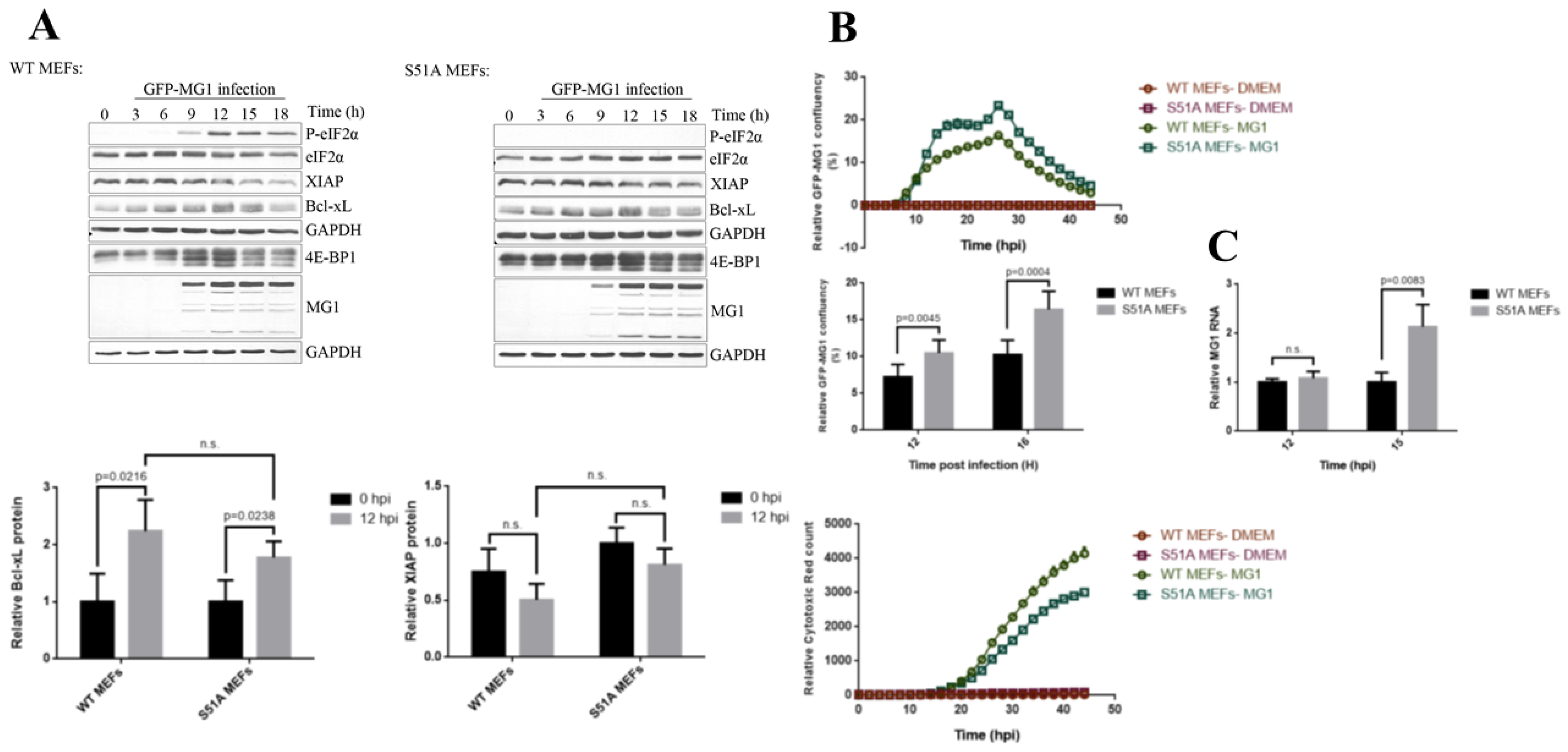
2.3. Phosphorylation of eIF2α Modulates the Repression of Host’s Protein Synthesis and MG1 Propagation
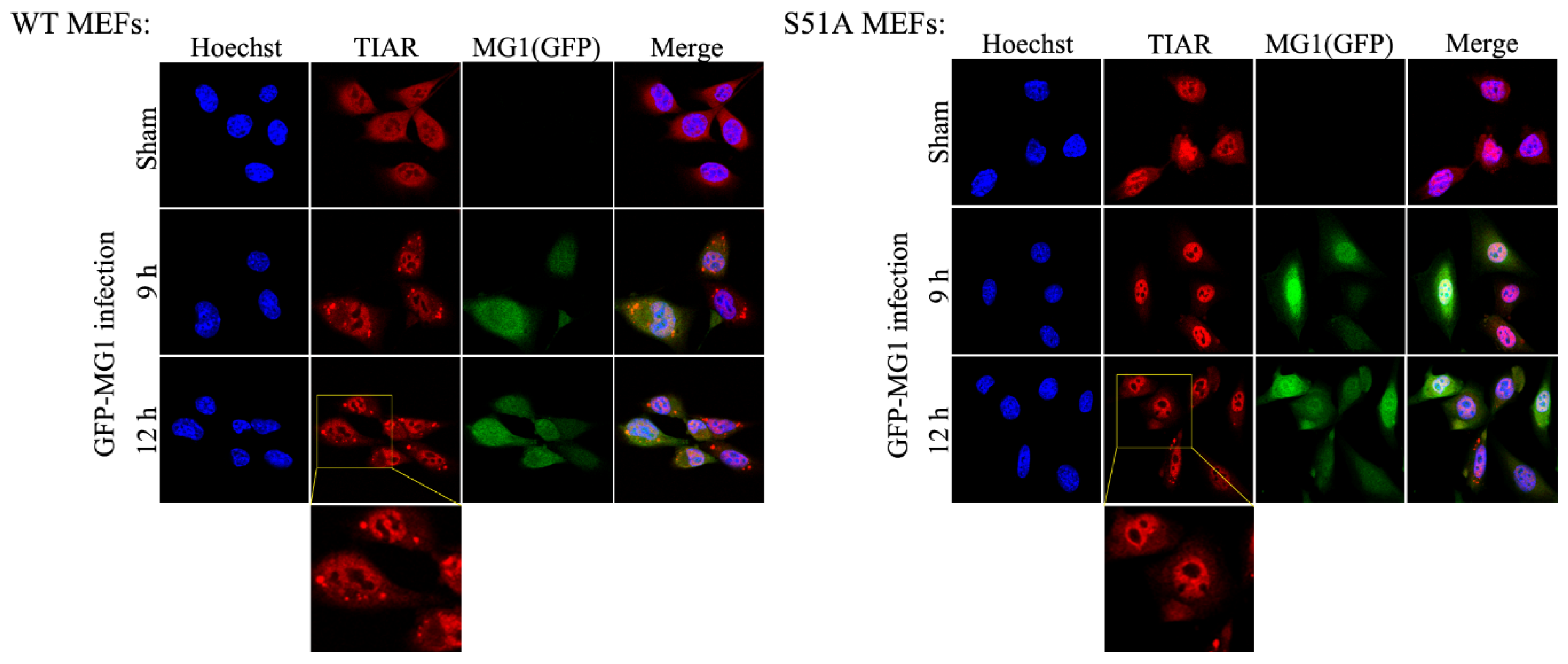
2.4. MG1-Induced Stress Granules Do Not Restrain Viral Propagation
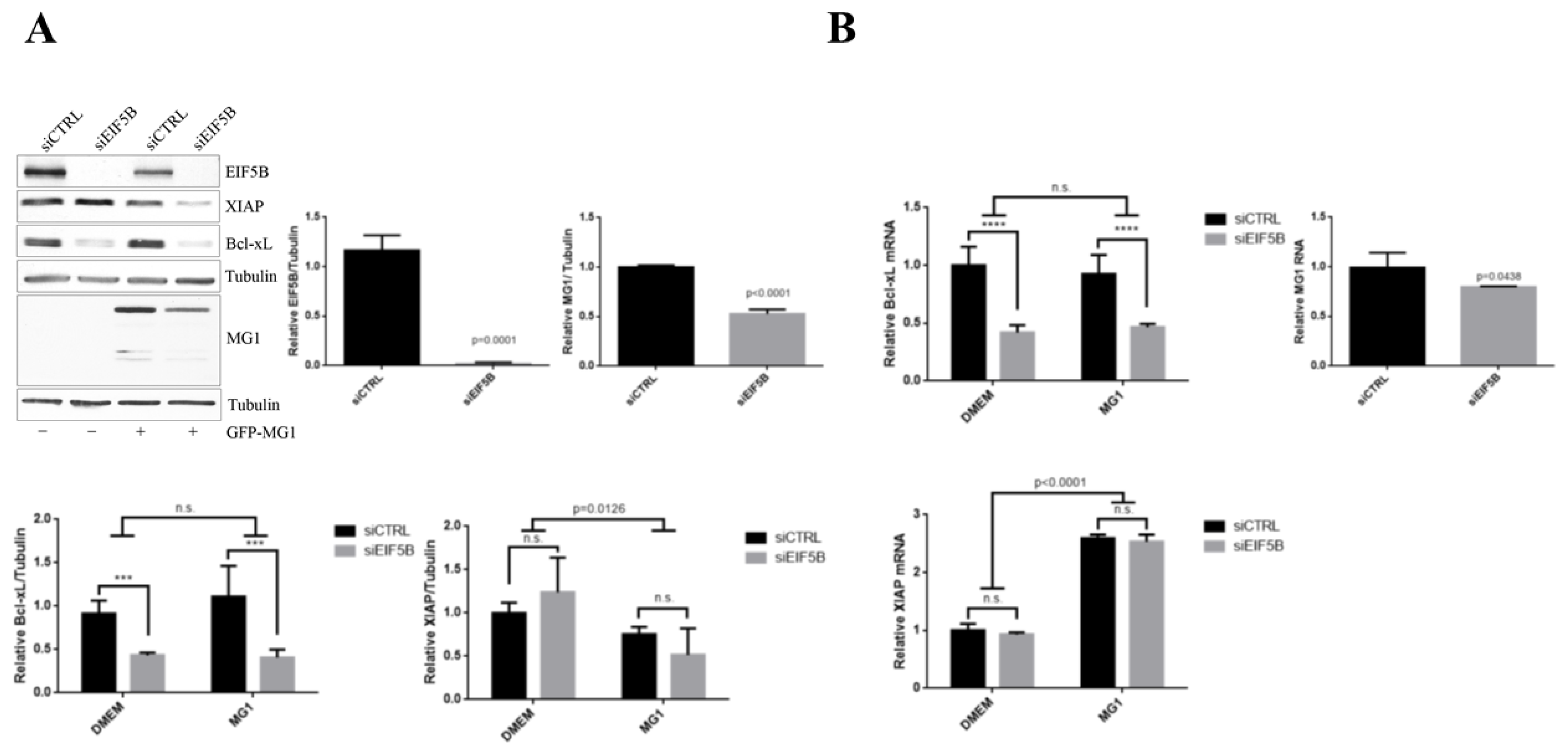
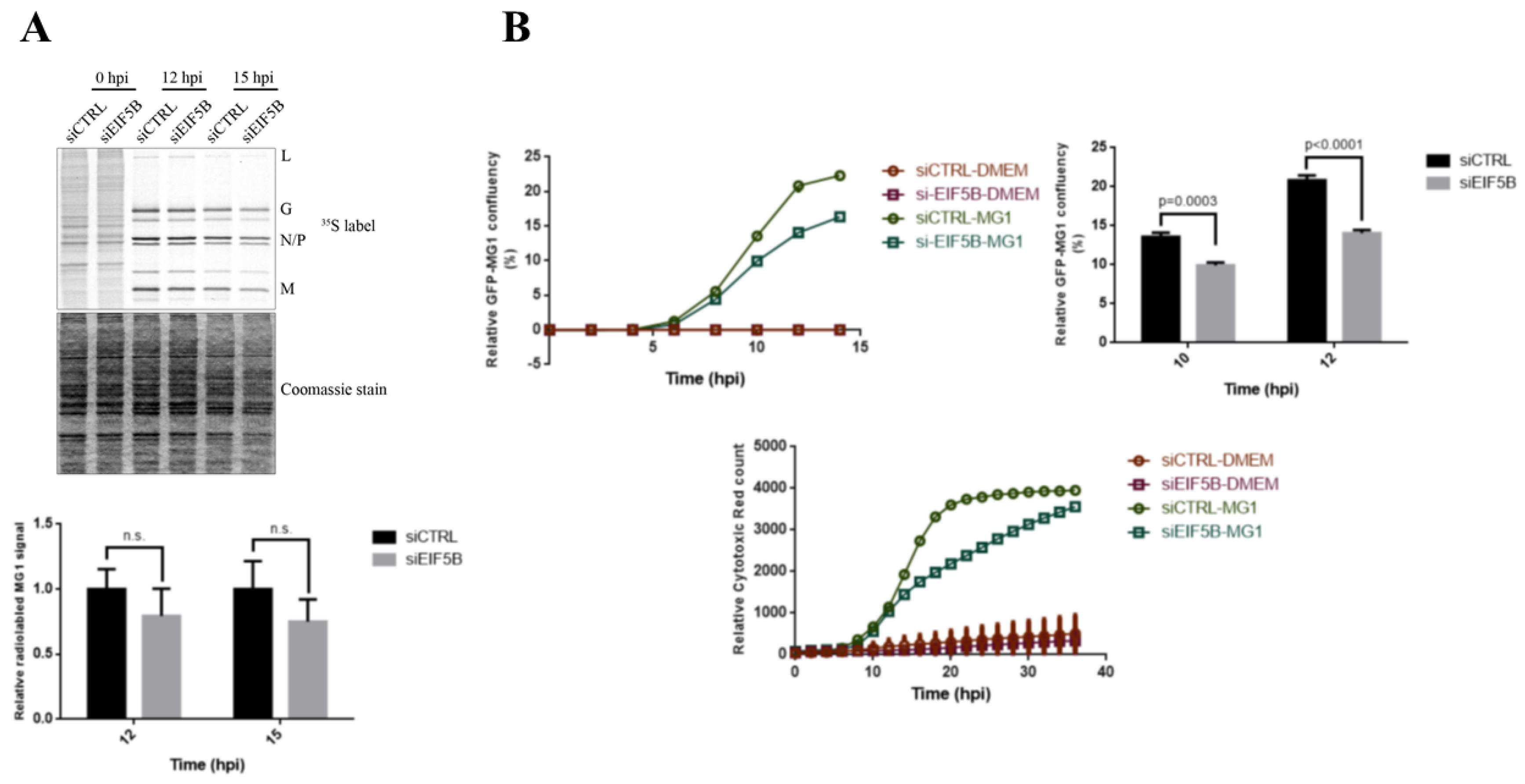
2.5. eIF5B Contributes to the Expression of Bcl-xL and MG1 during Viral Infection
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Expression Constructs and Transfection
4.2. Ribosome Profiling
4.3. Mapping and Analysis of Ribosome Profiling Data
4.4. Protein Extraction and Western Blot Analysis
4.5. [35S]-Methionine Labeling
4.6. Immunofluorescence and Confocal Microscopy
4.7. RNA Extraction and RT-qPCR
4.8. Kinetic Live Cell Imaging
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Travassos da Rosa, A.P.; Tesh, R.B.; Travassos da Rosa, J.F.; Herve, J.P.; Main, A.J., Jr. Carajas and Maraba viruses, two new vesiculoviruses isolated from phlebotomine sand flies in Brazil. Am. J. Trop. Med. Hyg. 1984, 33, 999–1006. [Google Scholar] [CrossRef]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol. Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tai, L.H.; Ilkow, C.S.; Alkayyal, A.A.; Ananth, A.A.; de Souza, C.T.; Wang, J.; Sahi, S.; Ly, L.; Lefebvre, C.; et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol. Ther. 2014, 22, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Hoeben, R.C. Non-human viruses developed as therapeutic agent for use in humans. Rev. Med. Virol. 2011, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Resseguier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Trask, S.D.; McDonald, S.M.; Patton, J.T. Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 2012, 10, 165–177. [Google Scholar] [CrossRef]
- Connor, J.H.; Lyles, D.S. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 2005, 280, 13512–13519. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. PKR; a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef]
- Raught, B.; Gingras, A.C. eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol. 1999, 31, 43–57. [Google Scholar] [CrossRef]
- Montero, H.; Garcia-Roman, R.; Mora, S.I. eIF4E as a control target for viruses. Viruses 2015, 7, 739. [Google Scholar] [CrossRef] [PubMed]
- Kapp, L.D.; Lorsch, J.R. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 2004, 73, 657–704. [Google Scholar] [CrossRef]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 7029–7036. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.H.; Lyles, D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 2002, 76, 10177–10187. [Google Scholar] [CrossRef]
- Koromilas, A.E. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Lorsch, J.R.; Dever, T.E. Molecular view of 43 S complex formation and start site selection in eukaryotic translation initiation. J. Biol. Chem. 2010, 285, 21203–21207. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Regulation of mRNA translation. Essays Biochem. 2001, 37, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Holcik, M. Could the eIF2alpha-Independent Translation Be the Achilles Heel of Cancer? Front. Oncol. 2015, 5, 264. [Google Scholar] [CrossRef]
- Taylor, D.R.; Lee, S.B.; Romano, P.R.; Marshak, D.R.; Hinnebusch, A.G.; Esteban, M.; Mathews, M.B. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol. Cell. Biol. 1996, 16, 6295–6302. [Google Scholar] [CrossRef]
- Lloyd, R.E. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip. Rev. RNA 2013, 4, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence That Ternary Complex (eIF2-GTP-tRNA(i)(Met))-Deficient Preinitiation Complexes Are Core Constituents of Mammalian Stress Granules. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eIF2, TIA-1 and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Dauber, B.; Pelletier, J.; Smiley, J.R. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 2011, 85, 5363–5373. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of Cytoplasmic mRNA Stress Granule Formation by a Viral Proteinase. Cell. Host Microbe. 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Lomakin, I.B.; Lee, J.H.; Choi, S.K.; Dever, T.E.; Hellen, C.U. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 2000, 403, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Kuhle, B.; Ficner, R. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J. 2014, 33, 1177–1191. [Google Scholar] [CrossRef]
- Terenin, I.M.; Dmitriev, S.E.; Andreev, D.E.; Shatsky, I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 2008, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Thakor, N.; Holcik, M. IRES-mediated translation of cellular messenger RNA operates in eIF2alpha- independent manner during stress. Nucleic Acids Res. 2012, 40, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; de Breyne, S.; Pisarev, A.V.; Abaeva, I.S.; Hellen, C.U. eIF2-dependent and eIF2-independent modes of initiation on the CSFV IRES: A common role of domain II. EMBO J. 2008, 27, 1060–1072. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hellen, C.U.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001, 15, 1593–1612. [Google Scholar] [CrossRef]
- Yoon, A.; Peng, G.; Brandenburg, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef]
- Bevilacqua, E.; Wang, X.; Majumder, M.; Gaccioli, F.; Yuan, C.L.; Wang, C.; Zhu, X.; Jordan, L.E.; Scheuner, D.; Kaufman, R.J.; et al. eIF2alpha phosphorylation tips the balance to apoptosis during osmotic stress. J. Biol. Chem. 2010, 285, 17098–17111. [Google Scholar] [CrossRef] [PubMed]
- Durie, D.; Hatzoglou, M.; Chakraborty, P.; Holcik, M. HuR controls mitochondrial morphology through the regulation of BclxL translation. Translation 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Jafarnejad, S.M.; Chapat, C.; Matta-Camacho, E.; Gelbart, I.A.; Hesketh, G.G.; Arguello, M.; Garzia, A.; Kim, S.H.; Attig, J.; Shapiro, M.; et al. Translational control of ERK signaling through miRNA/4EHP-directed silencing. Elife 2018, 7, 35034. [Google Scholar] [CrossRef]
- Hoang, H.D.; Graber, T.E.; Alain, T. Battling for Ribosomes: Translational Control at the Forefront of the Antiviral Response. J. Mol. Biol. 2018, 430, 1965–1992. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Dinh, P.X.; Beura, L.K.; Das, P.B.; Panda, D.; Das, A.; Pattnaik, A.K. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J. Virol. 2013, 87, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Centrella, M.; Lucas-Lenard, J. Regulation of protein synthesis in vesicular stomatitis virus-infected mouse L-929 cells by decreased protein synthesis initiation factor 2 activity. J. Virol. 1982, 41, 781–791. [Google Scholar] [PubMed]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol 1999, 1, 190–192. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.H.; Vakifahmetoglu-Norberg, H.; Yuan, J. Integration of apoptosis and metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Korneluk, R.G. XIAP, the guardian angel. Nat. Rev. Mol. Cell Biol. 2001, 2, 550–556. [Google Scholar] [CrossRef]
- Holcik, M. Translational upregulation of the X-linked inhibitor of apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-Mediated Inhibition of Caspase-9. Mol. Cell 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Cuconati, A.; White, E. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev. 2002, 16, 2465–2478. [Google Scholar] [CrossRef]
- Allam, H.; Ali, N. Initiation factor eIF2-independent mode of c-Src mRNA translation occurs via an internal ribosome entry site. J. Biol. Chem. 2010, 285, 5713–5725. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yang, Y.; Hoang, B.; Bardeleben, C.; Holmes, B.; Gera, J.; Lichtenstein, A. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene 2016, 35, 1015–1024. [Google Scholar] [CrossRef]
- Yamamoto, H.; Unbehaun, A.; Loerke, J.; Behrmann, E.; Collier, M.; Burger, J.; Mielke, T.; Spahn, C.M. Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat Struct Mol. Biol. 2014, 21, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A. FASTQ/A Short-Reads Pre-Processing Tools. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 1 January 2018).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Lefebvre, C.A.; Hicks, K.; Korneluk, R.G. Cloning and characterization of the rat homologues of the Inhibitor of Apoptosis protein 1, 2 and 3 genes. BMC Genom. 2002, 3, 5. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanzadeh, G.; Naing, T.; Graber, T.; Jafarnejad, S.M.; Stojdl, D.F.; Alain, T.; Holcik, M. Characterizing Cellular Responses During Oncolytic Maraba Virus Infection. Int. J. Mol. Sci. 2019, 20, 580. https://doi.org/10.3390/ijms20030580
Hassanzadeh G, Naing T, Graber T, Jafarnejad SM, Stojdl DF, Alain T, Holcik M. Characterizing Cellular Responses During Oncolytic Maraba Virus Infection. International Journal of Molecular Sciences. 2019; 20(3):580. https://doi.org/10.3390/ijms20030580
Chicago/Turabian StyleHassanzadeh, Golnoush, Thet Naing, Tyson Graber, Seyed Mehdi Jafarnejad, David F. Stojdl, Tommy Alain, and Martin Holcik. 2019. "Characterizing Cellular Responses During Oncolytic Maraba Virus Infection" International Journal of Molecular Sciences 20, no. 3: 580. https://doi.org/10.3390/ijms20030580
APA StyleHassanzadeh, G., Naing, T., Graber, T., Jafarnejad, S. M., Stojdl, D. F., Alain, T., & Holcik, M. (2019). Characterizing Cellular Responses During Oncolytic Maraba Virus Infection. International Journal of Molecular Sciences, 20(3), 580. https://doi.org/10.3390/ijms20030580