Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery

 , , ,
, , , 
Abstract
1. Introduction
2. Results
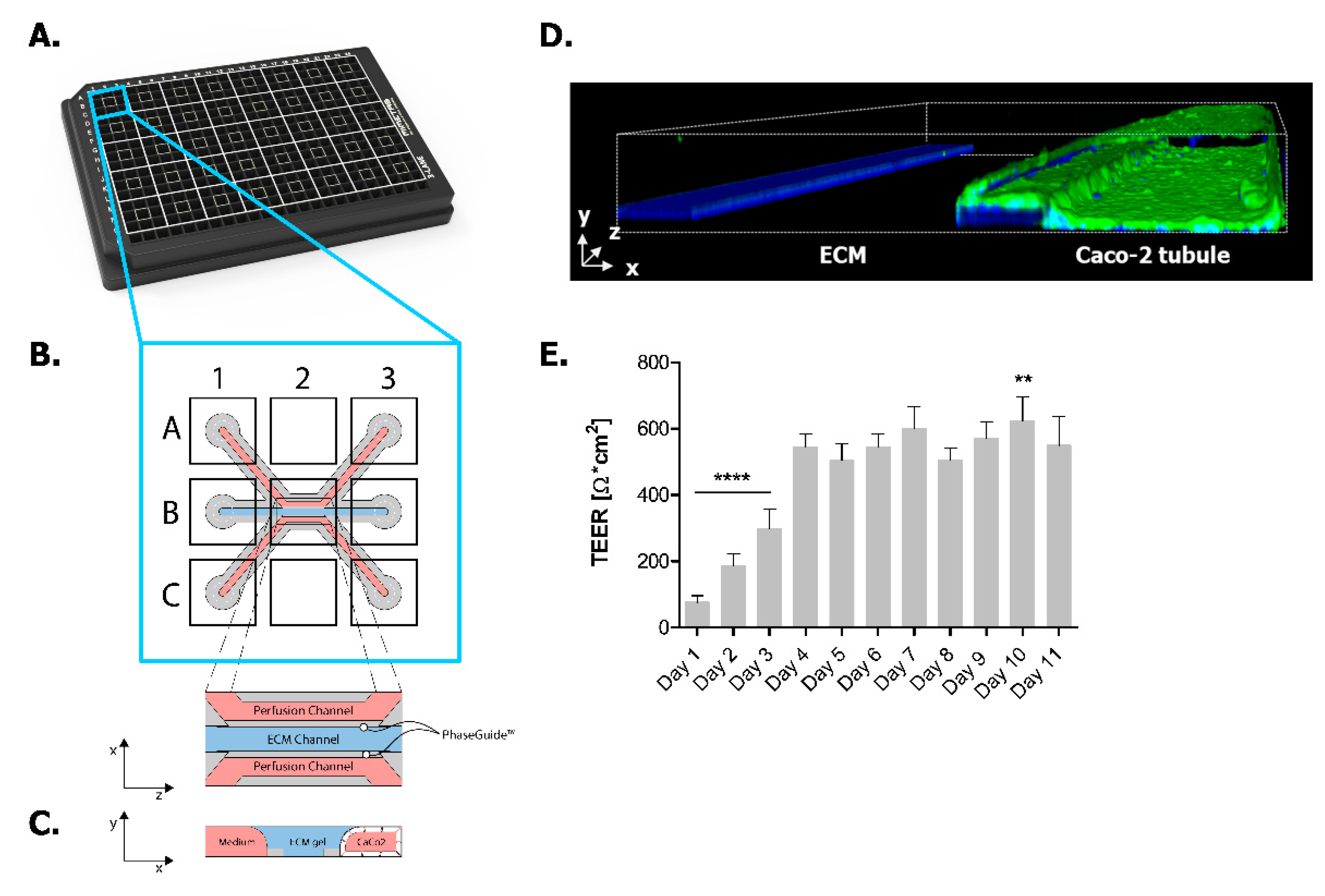
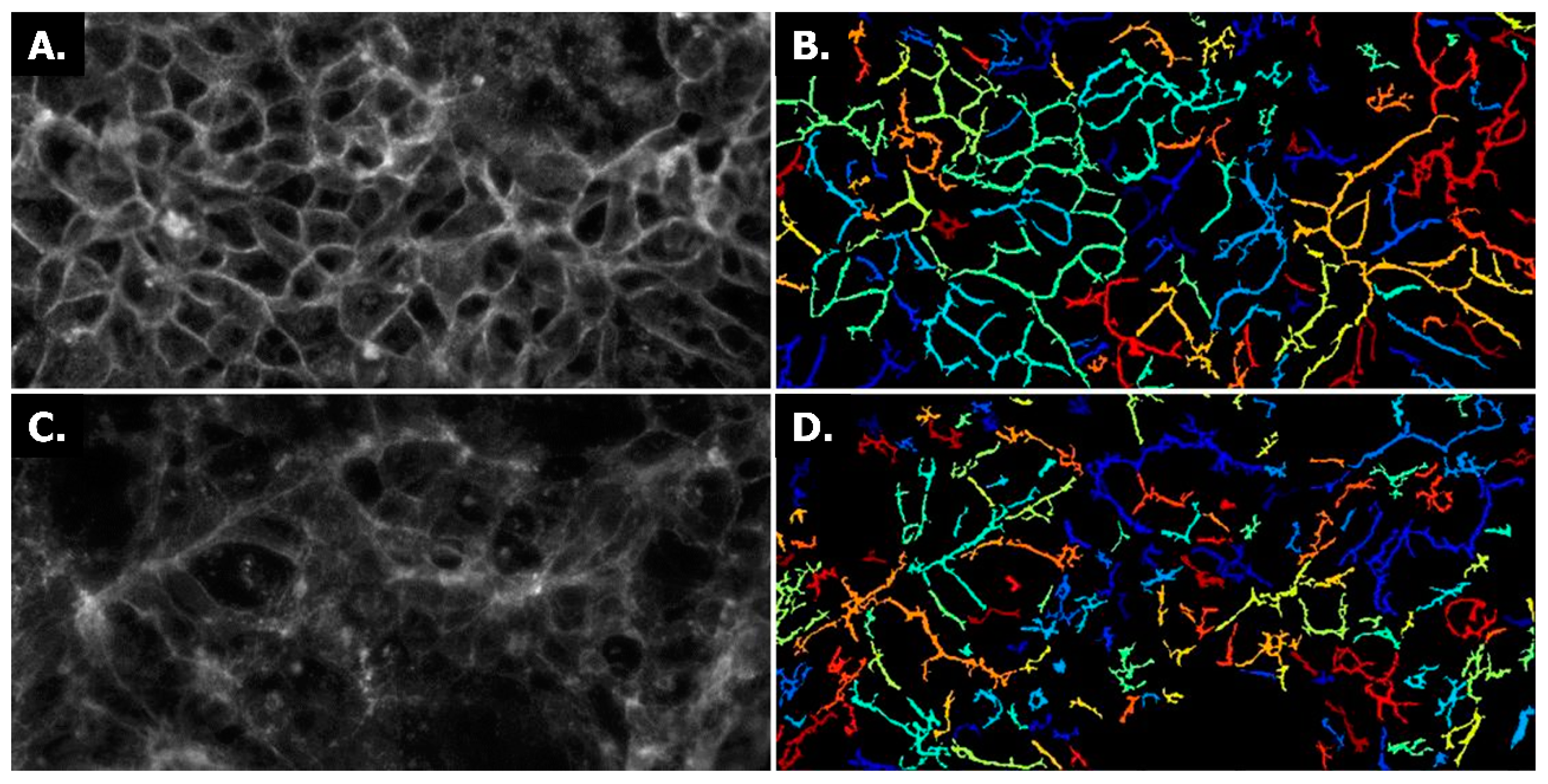
2.1. Establishing Leak-Tight 3D Caco-2 Tubules Controlled by TEER Measurements
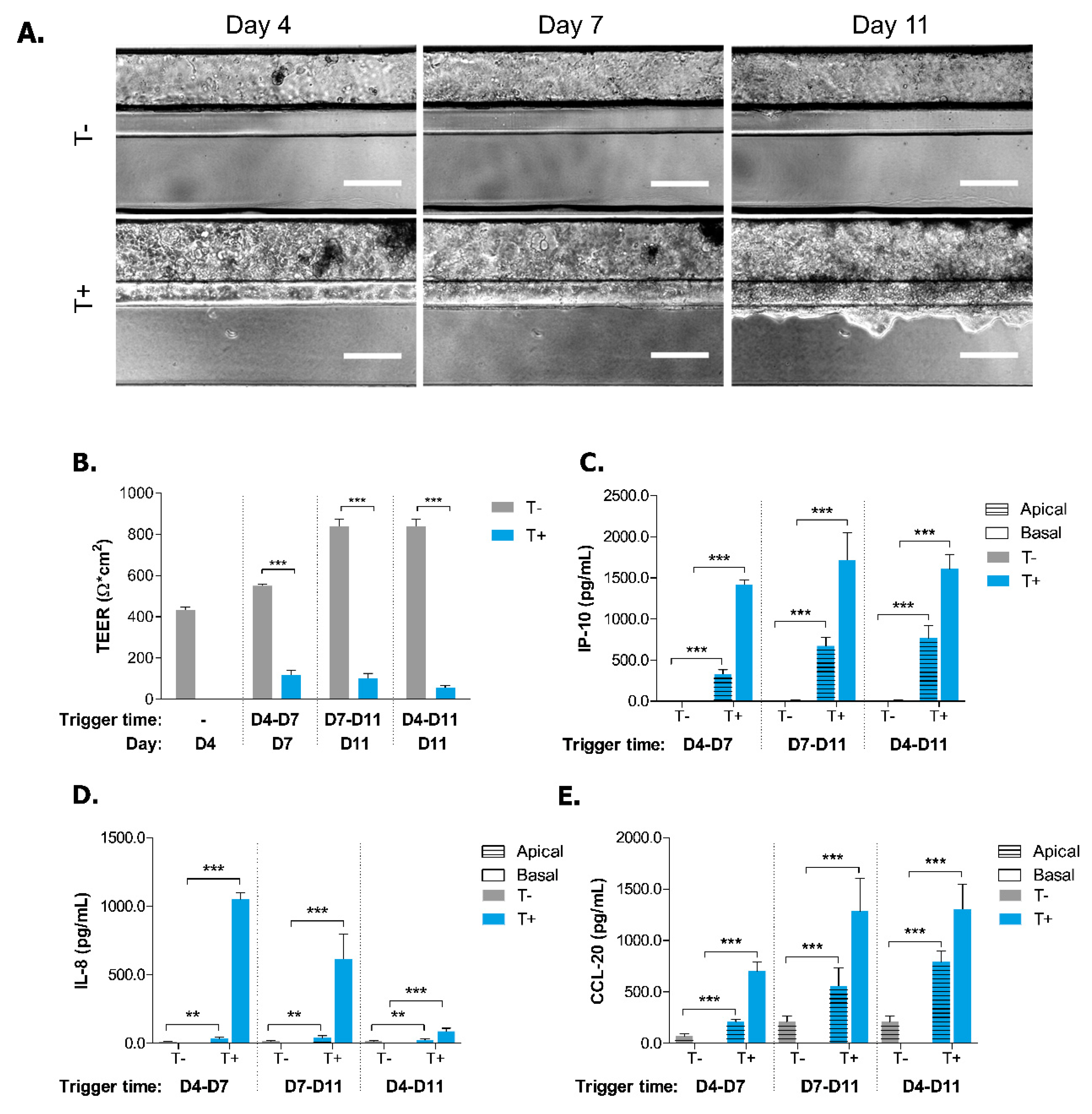
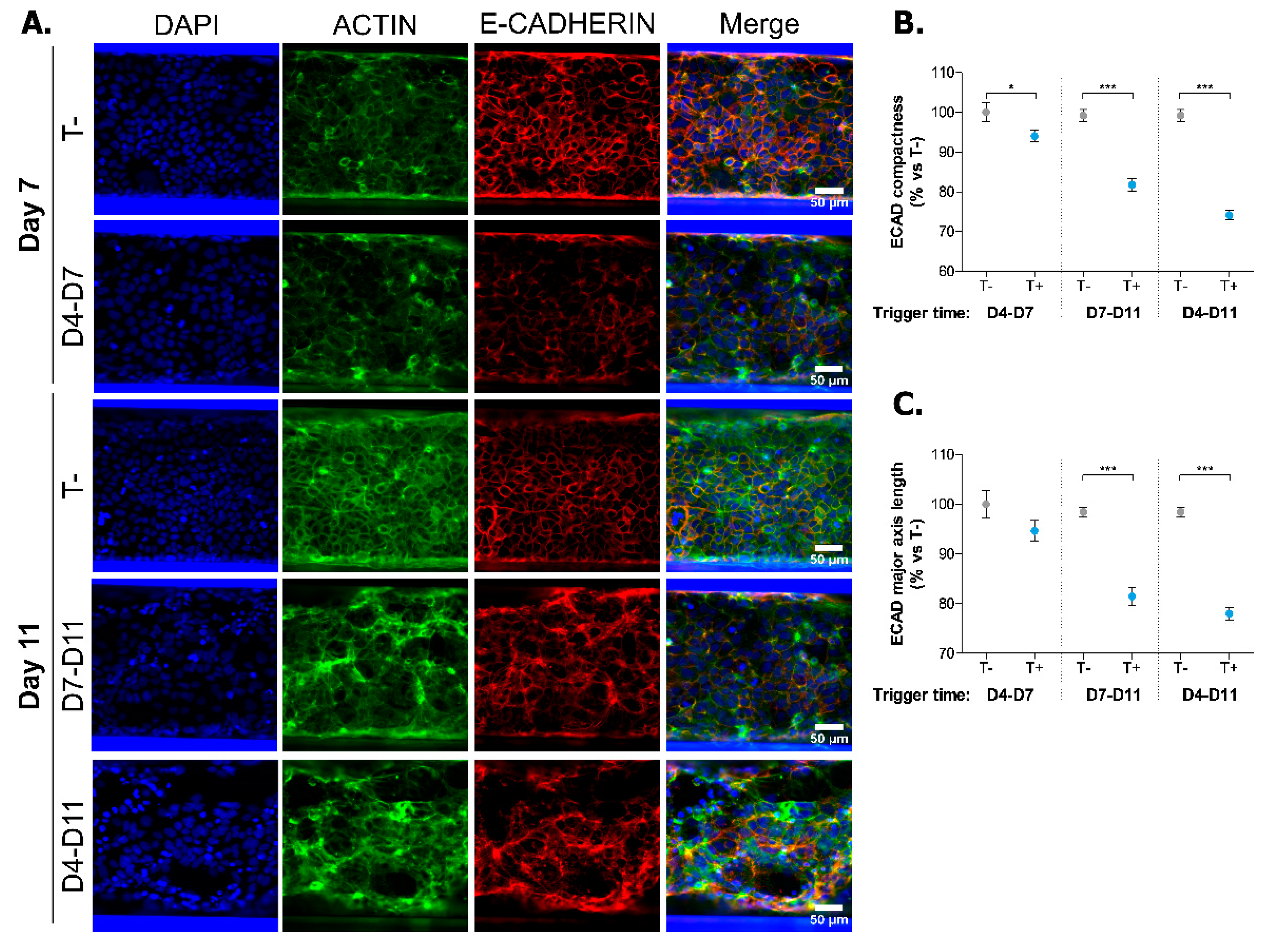
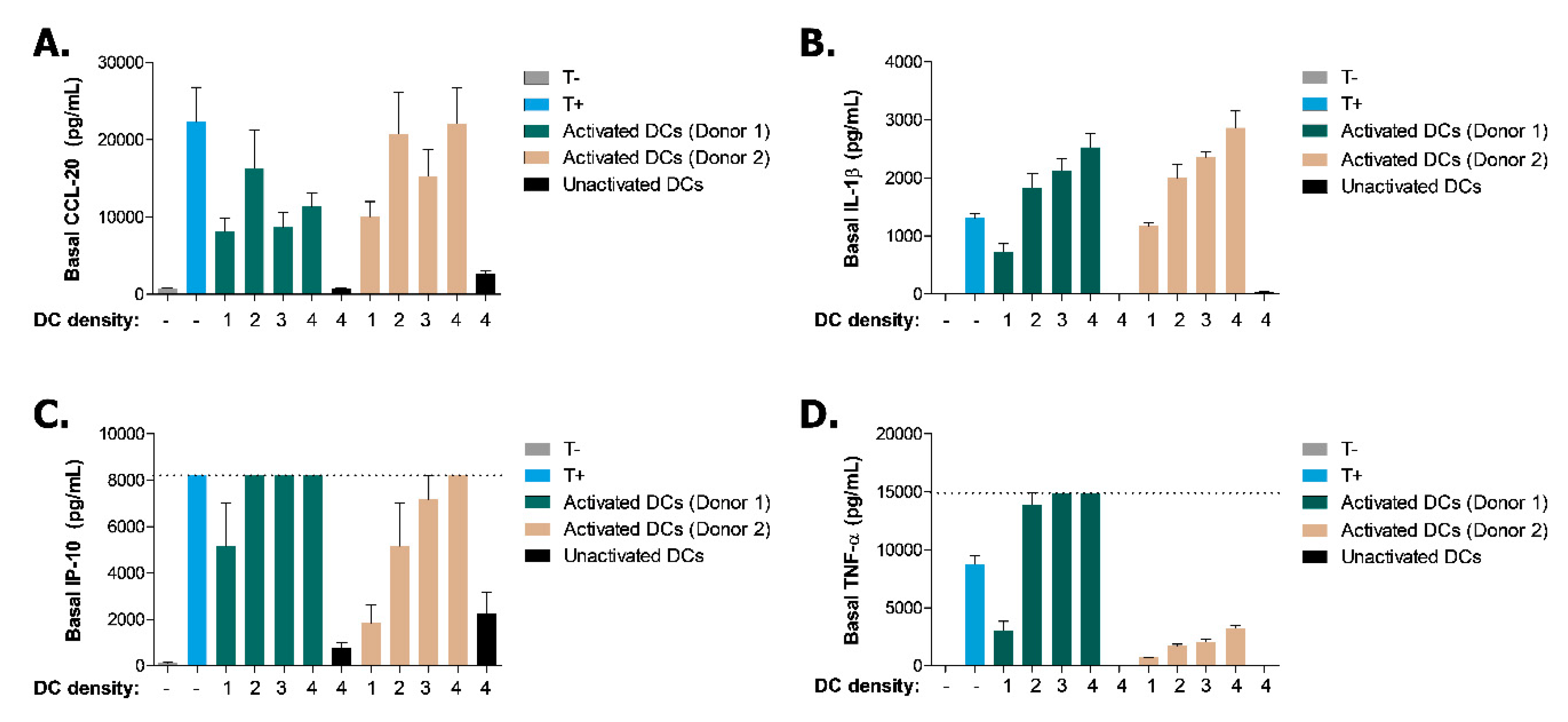
2.2. Induction of Inflammatory State in Caco-2 Tubules
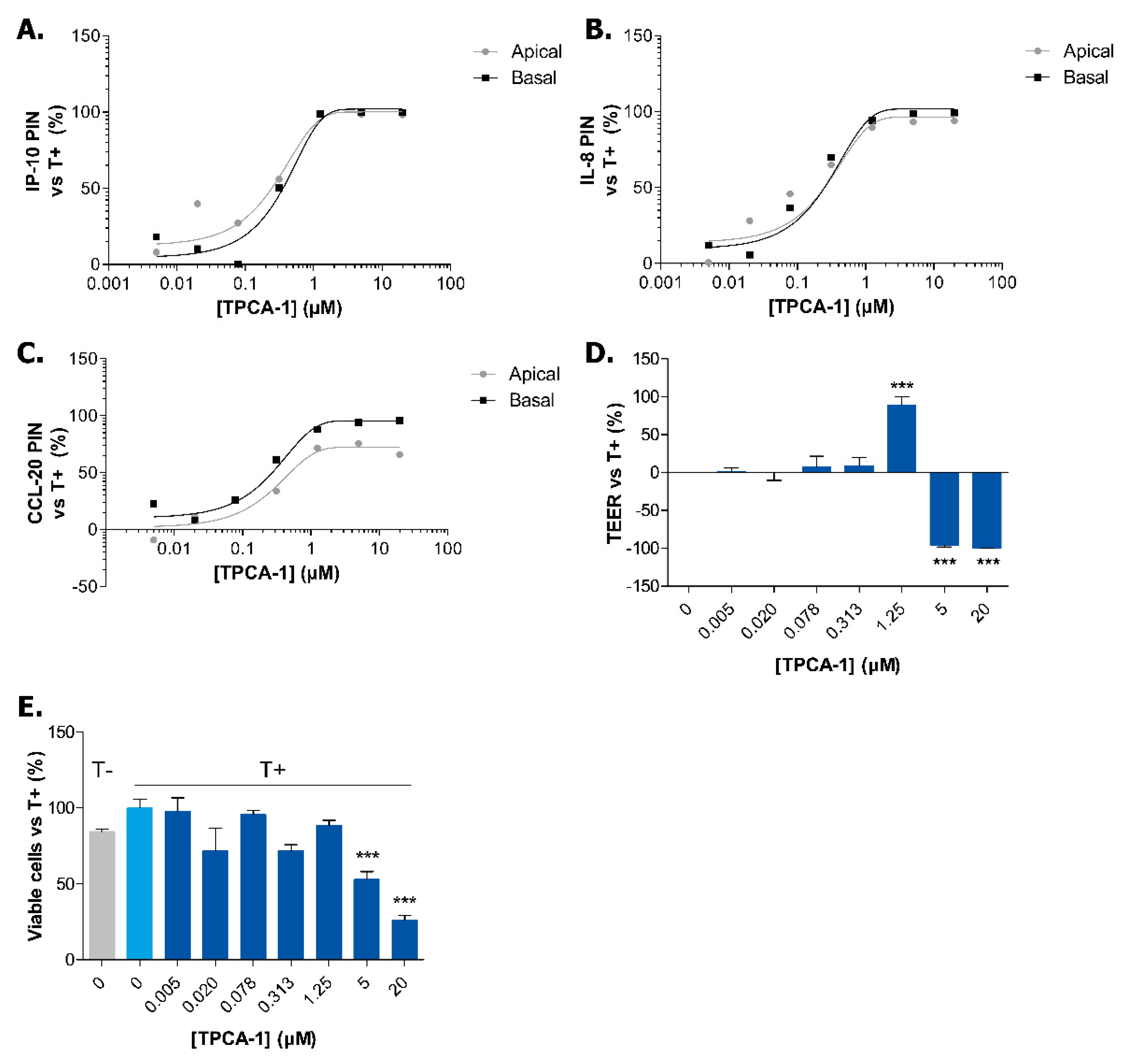
2.3. Exposure to TPCA-1 Prevent the Inflammatory State of Caco-2 Tubules
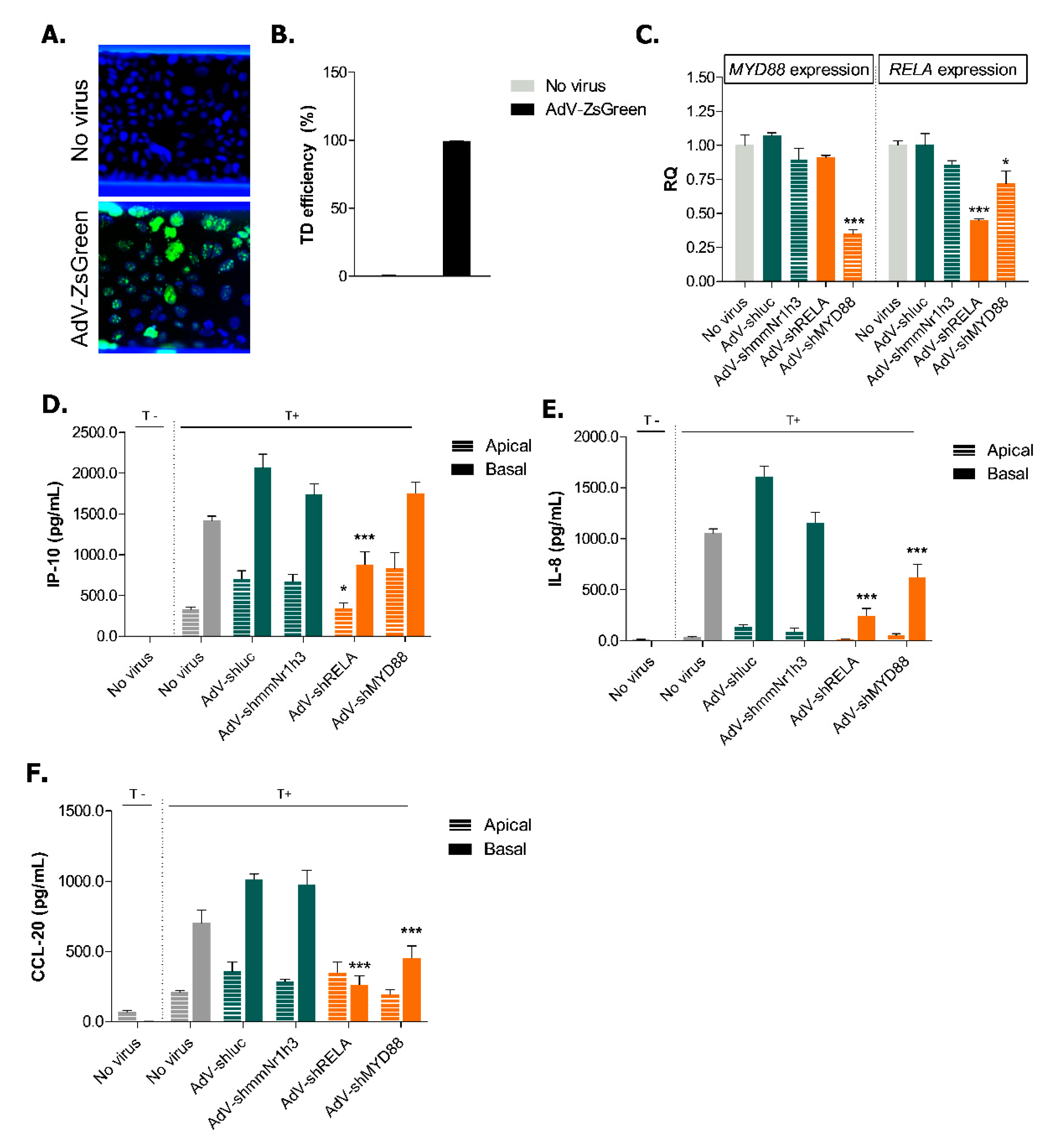
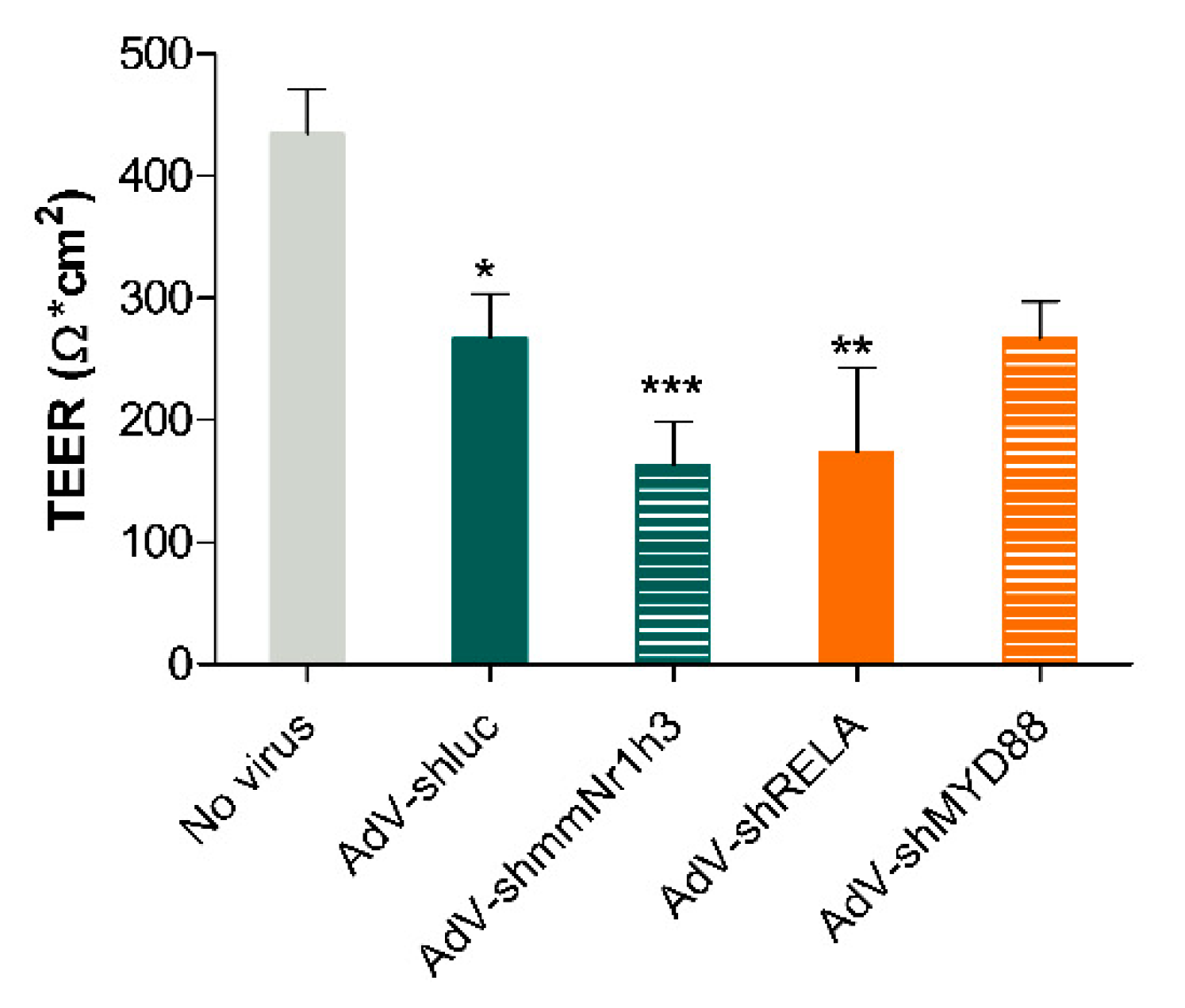
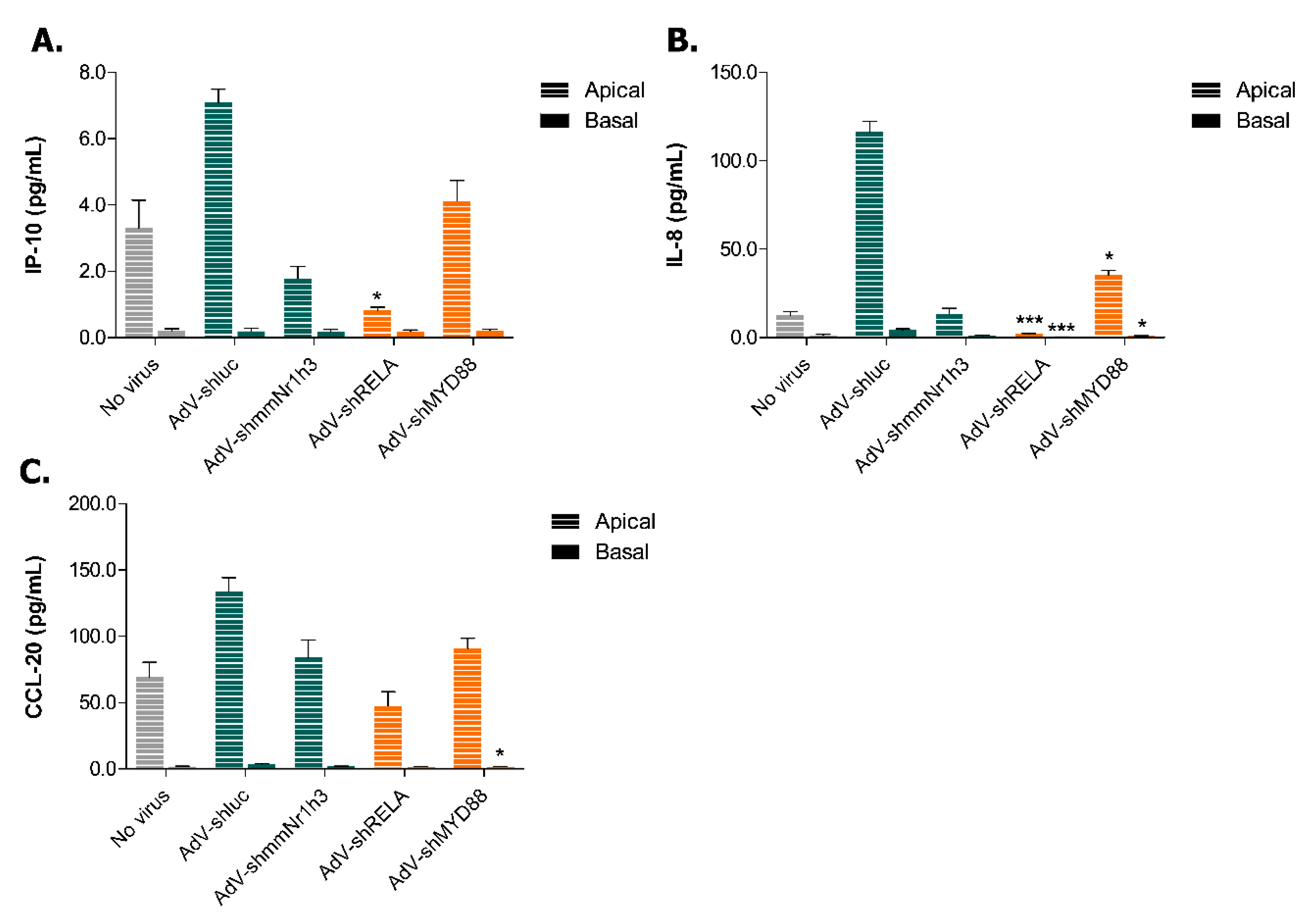
2.4. Adenoviral Knockdown of Inflammatory Effectors Prevents IBD-like Phenotype in Caco-2 Tubules
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cells
4.3. OrganoPlate Seeding and Tubule Formation
4.4. Triggering of IBD-like Conditions
4.5. Cytokine Secretion of Caco-2 Cells
4.6. TEER Measurements
4.7. Immunohistochemistry
4.8. Adenoviral Preparation
4.9. Adenoviral Transduction
4.10. Transduction Efficiency
4.11. Quantitative PCR
4.12. Compound Exposure
4.13. Viability Assay
4.14. Statistics and Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AdV | Adenovirus |
| AJ | Adherens junction |
| CD | Crohn’s disease |
| DC | Dendritic cell |
| ECM | Extracellular matrix |
| IBD | Inflammatory bowel disease |
| IEC | Intestinal epithelial cells |
| iPSC | Induced pluripotent stem cell |
| NEAA | Non-essential amino acids |
| PBMC | Peripheral blood mononuclear cells |
| RQ | Relative quantification |
| TEER | Transepithelial electrical resistance |
| TJ | Tight junction |
| UC | Ulcerative colitis |
Appendix A. Supplementary Methods
Appendix A.1. Cells
Appendix A.2. Optimization of the Immune-Relevant Trigger
Appendix A.3. Selection of Viruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insert Name | Target Sequence |
|---|---|
| aveGFP_a | GCCACAACGTCTATATCAT |
| ffluc_a | GCAGAAGCTATGAAACGAT |
| ffluc_b | GCATGCCAGAGATCCTATT |
| luc_a * | GGTTACCTAAGGGTGTGGC |
| mmNr1h3_a * | CACACATATGTGGAGGCCC |
| Insert Name | Target Sequence |
|---|---|
| MYD88_a | GGAACAGACAAACTATCGA |
| MYD88_b | GGTTGTCTCTGATGATTAC |
| MYD88_c | GACTTCCAGACCAAATTTG |
| MYD88_d * | GGTTCATCACTGTCTGCGA |
| MYD88_e | CTGCTCTCAACATGCGAGT |
| RELA_a * | GATTGAGGAGAAACGTAAA |
| RELA_b | GTACCCTGAGGCTATAACT |
Appendix B. Supplementary Data






References
- De Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L. The burden of inflammatory bowel disease in Europe. J. Crohn’s Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef] [PubMed]
- De Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kanai, T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.; Coskun, M.; Soendergaard, C.; Salem, M.; Nielsen, O.H. Inflammatory pathways of importance for management of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. IBD pathogenesis in 2014: Molecular pathways controlling barrier function in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.J.; Lobo, A.J.; Travis, S.P.L.; IBD Section, British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2004, 53 (Suppl. 5), V1–V16. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A. Animal Models of Inflammatory Bowel Disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [PubMed]
- Tsilingiri, K.; Barbosa, T.; Penna, G.; Caprioli, F.; Sonzogni, A.; Viale, G.; Rescigno, M. Probiotic and postbiotic activity in health and disease: Comparison on a novel polarised ex-vivo organ culture model. Gut 2012, 61, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Middendorp, S.; Wiegerinck, C.L.; Witte, M.; Teunissen, H.; Meddens, C.A.; Cuppen, E.; Clevers, H.; Nieuwenhuis, E.E.S. Many Inflammatory Bowel Disease Risk Loci Include Regions That Regulate Gene Expression in Immune Cells and the Intestinal Epithelium. Gastroenterology 2014, 146, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.M.; Philpott, D.J.; Perdue, M.H. Review article: In vitro models in inflammatory bowel disease research—A critical review. Aliment. Pharmacol. Ther. 1997, 11 (Suppl. 3), 70–80. [Google Scholar] [CrossRef] [PubMed]
- Eri, R.; McGuckin, M.A.; Wadley, R. T cell transfer model of colitis: A great tool to assess the contribution of T cells in chronic intestinal inflammation. Methods Mol. Biol. 2012, 844, 261–275. [Google Scholar] [PubMed]
- Roy, U.; Galvez, E.J.C.; Iljazovic, A.; Lesker, T.R.; Blazejewski, A.J.; Pils, M.C.; Heise, U.; Huber, S.; Flavell, R.A.; Strowig, T. Distinct Microbial Communities Trigger Colitis Development upon Intestinal Barrier Damage via Innate or Adaptive Immune Cells. Cell Rep. 2017, 21, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712. [Google Scholar] [CrossRef] [PubMed]
- Fritschfredin, M.; Vidal, A.; Utkovic, H.; Gotlind, Y.; Willen, R.; Jansson, L.; Hultgrenhornquist, E.; Melgar, S. The application and relevance of ex vivo culture systems for assessment of IBD treatment in murine models of colitis. Pharmacol. Res. 2008, 58, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Vadstrup, K.; Galsgaard, E.D.; Gerwien, J.; Vester-Andersen, M.K.; Pedersen, J.S.; Rasmussen, J.; Neermark, S.; Kiszka-Kanowitz, M.; Jensen, T.; Bendtsen, F. Validation and Optimization of an Ex Vivo Assay of Intestinal Mucosal Biopsies in Crohn’s Disease: Reflects Inflammation and Drug Effects. PLoS ONE 2016, 11, e0155335. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, A.R.; Conradi, R.A.; Burton, P.S. Caco-2 Cell Monolayers as a Model for Drug Transport Across the Intestinal Mucosa. Pharm. Res. 1990, 7, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018, 8, 2871. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.-H.; Kim, H.J. Human gut-on-a-chip technology: Will this revolutionize our understanding of IBD and future treatments? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 883–885. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toepke, M.W.; Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 2006, 6, 1484. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Trietsch, S.J.; Naumovska, E.; Kurek, D.; Setyawati, M.C.; Vormann, M.K.; Wilschut, K.J.; Lanz, H.L.; Nicolas, A.; Ng, C.P.; Joore, J.; et al. Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nat. Commun. 2017, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Bertholet, S.; Moutaftsi, M.; Guderian, J.A.; Windish, H.P.; Baldwin, S.L.; Laughlin, E.M.; Duthie, M.S.; Fox, C.B.; Carter, D.; et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS ONE 2011, 6, e16333. [Google Scholar] [CrossRef] [PubMed]
- De Castro, C.P.; Jones, S.A.; Ni Cheallaigh, C.; Hearnden, C.A.; Williams, L.; Winter, J.; Lavelle, E.C.; Mills, K.H.G.; Harris, J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J. Immunol. 2012, 189, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Gad, M.; Ravn, P.; Soborg, D.A.; Lund-Jensen, K.; Ouwehand, A.C.; Jensen, S.S. Regulation of the IL-10/IL-12 axis in human dendritic cells with probiotic bacteria. FEMS Immunol. Med. Microbiol. 2011, 63, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Callahan, J.F.; Bolognese, B.J.; Li, Y.H.; Carlson, K.; Davis, T.G.; Mellor, G.W.; Evans, C.; Roshak, A.K. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB Kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and. J. Pharmacol. Exp. Ther. 2005, 312, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Balakin, K.V.; Lavrovsky, Y. Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini Rev. Med. Chem. 2011, 11, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G.A. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 2011, 11, 1596. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Van De Walle, J.; Hendrickx, A.; Romier, B.; Larondelle, Y.; Schneider, Y.-J. Inflammatory parameters in Caco-2 cells: Effect of stimuli nature, concentration, combination and cell differentiation. Toxicol. In Vitro 2010, 24, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Coskun, M. Intestinal epithelium in inflammatory bowel disease. Front. Med. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Karayiannakis, A.J.; Syrigos, K.N.; Efstathiou, J.; Valizadeh, A.; Noda, M.; Playford, R.J.; Kmiot, W.; Pignatelli, M. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J. Pathol. 1998, 185, 413–418. [Google Scholar] [CrossRef]
- Zbar, A.P.; Simopoulos, C.; Karayiannakis, A.J. Cadherins: An integral role in inflammatory bowel disease and mucosal restitution. J. Gastroenterol. 2004, 39, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Hanby, A.M.; Chinery, R.; Poulsom, R.; Playford, R.J.; Pignatelli, M. Downregulation of E-cadherin in the reparative epithelium of the human gastrointestinal tract. Am. J. Pathol. 1996, 148, 723–729. [Google Scholar] [PubMed]
- Lawrence, T. The Nuclear Factor NF- B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Tran, R.; Myers, D.R.; Denning, G.; Shields, J.E.; Lytle, A.M.; Alrowais, H.; Qiu, Y.; Sakurai, Y.; Li, W.C.; Brand, O.; et al. Microfluidic Transduction Harnesses Mass Transport Principles to Enhance Gene Transfer Efficiency. Mol. Ther. 2017, 25, 2372–2382. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Shankar, N. Roles of TLR/MyD88/MAPK/NF-κB Signaling Pathways in the Regulation of Phagocytosis and Proinflammatory Cytokine Expression in Response to E. faecalis Infection. PLoS ONE 2015, 10, e0136947. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Adams, C.P.; Brantner, V.V. Spending on new drug development. Health Econ. 2010, 19, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Franzen, N.; van Harten, W.H.; Retèl, V.P.; Loskill, P.; van den Eijnden-van Raaij, J.; IJzerman, M. Impact of organ-on-a-chip technology on pharmaceutical R&D costs. Drug Discov. Today 2019, 24, 1720–1724. [Google Scholar] [PubMed]
- Maznah, I. The use of Caco-2 cells as an in vitro method to study bioavailability of iron. Malays. J. Nutr. 1999, 5, 31–45. [Google Scholar] [PubMed]
- Hellinger, É.; Veszelka, S.; Tóth, A.E.; Walter, F.; Kittel, Á.; Bakk, M.L.; Tihanyi, K.; Háda, V.; Nakagawa, S.; Duy, T.D.H.; et al. Comparison of brain capillary endothelial cell-based and epithelial (MDCK-MDR1, Caco-2, and VB-Caco-2) cell-based surrogate blood–brain barrier penetration models. Eur. J. Pharm. Biopharm. 2012, 82, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Odijk, M.; Van Der Meer, A.D.; Levner, D.; Kim, H.J.; Van Der Helm, M.W.; Segerink, L.I.; Frimat, J.P.; Hamilton, G.A.; Ingber, D.E.; Van Den Berg, A. Measuring direct current trans-epithelial electrical resistance in organ-on-a-chip microsystems. Lab Chip 2015, 15, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Ligumsky, M.; Simon, P.L.; Karmeli, F.; Rachmilewitz, D. Role of interleukin 1 in inflammatory bowel disease--enhanced production during active disease. Gut 1990, 31, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chaudhary, R.; Carpani, M.; Playford, R. Interfering with interferons in inflammatory bowel disease. Gut 2005, 55, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Dionne, S.; Hiscott, J.; D’Agata, I.; Duhaime, A.; Seidman, E.G. Quantitative PCR Analysis of TNF-α and IL-1β mRNA Levels in Pediatric IBD Mucosal Biopsies. Dig. Dis. Sci. 1997, 42, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, R.; Koide, T.; Tokoro, C.; Yamamoto, T.; Godai, T.; Morohashi, T.; Fujita, Y.; Takahashi, D.; Kawana, I.; Suzuki, S.; et al. Quantitive Cytokine mRNA Expression Profiles in the Colonic Mucosa of Patients with Steroid Naïve Ulcerative Colitis During Active and Quiescent Disease. Inflamm. Bowel Dis. 2009, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Onyiah, J.C.; Colgan, S.P. Cytokine responses and epithelial function in the intestinal mucosa. Cell. Mol. Life Sci. 2016, 73, 4203–4212. [Google Scholar] [CrossRef] [PubMed]
- Panja, A.; Goldberg, S.; Eckmann, L.; Krishen, P.; Mayer, L. The regulation and functional consequence of proinflammatory cytokine binding on human intestinal epithelial cells. J. Immunol. 1998, 161, 3675–3684. [Google Scholar] [PubMed]
- Muise, A.M.; Walters, T.D.; Glowacka, W.K.; Griffiths, A.M.; Ngan, B.-Y.; Lan, H.; Xu, W.; Silverberg, M.S.; Rotin, D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut 2009, 58, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, L.; Jung, H.C.; Schurer-Maly, C.; Panja, A.; Morzycka-Wroblewska, E.; Kagnoff, M.F. Differential cytokine expression by human intestinal epithelial cell lines: Regulated expression of interleukin 8. Gastroenterology 1993, 105, 1689–1697. [Google Scholar] [CrossRef]
- Workman, M.J.; Gleeson, J.P.; Troisi, E.J.; Estrada, H.Q.; Kerns, S.J.; Hinojosa, C.D.; Hamilton, G.A.; Targan, S.R.; Svendsen, C.N.; Barrett, R.J. Enhanced Utilization of Induced Pluripotent Stem Cell–Derived Human Intestinal Organoids Using Microengineered Chips. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 669–677.e2. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory bowel disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Arts, G.-J.; Langemeijer, E.; Tissingh, R.; Ma, L.; Pavliska, H.; Dokic, K.; Dooijes, R.; Mesic, E.; Clasen, R.; Michiels, F.; et al. Adenoviral vectors expressing siRNAs for discovery and validation of gene function. Genome Res. 2003, 13, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Michiels, F.; van Es, H.; van Rompaey, L.; Merchiers, P.; Francken, B.; Pittois, K.; van der Schueren, J.; Brys, R.; Vandersmissen, J.; Beirinckx, F.; et al. Arrayed adenoviral expression libraries for functional screening. Nat. Biotechnol. 2002, 20, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Rippmann, J.F.; Schoelch, C.; Nolte, T.; Pavliska, H.; van Marle, A.; van Es, H.; Prestle, J. Improved lipid profile through liver-specific knockdown of liver X receptor alpha in KKAy diabetic mice. J. Lipid Res. 2009, 50, 22–31. [Google Scholar] [CrossRef] [PubMed]





| Insert Name | Target Sequence |
|---|---|
| AdV-shluc | GGTTACCTAAGGGTGTGGC |
| AdV_shmmNr1h3 | CACACATATGTGGAGGCCC |
| AdV_shMYD88 | GGTTCATCACTGTCTGCGA |
| AdV_shRELA | GATTGAGGAGAAACGTAAA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaurivage, C.; Naumovska, E.; Chang, Y.X.; Elstak, E.D.; Nicolas, A.; Wouters, H.; van Moolenbroek, G.; Lanz, H.L.; Trietsch, S.J.; Joore, J.; et al. Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery. Int. J. Mol. Sci. 2019, 20, 5661. https://doi.org/10.3390/ijms20225661
Beaurivage C, Naumovska E, Chang YX, Elstak ED, Nicolas A, Wouters H, van Moolenbroek G, Lanz HL, Trietsch SJ, Joore J, et al. Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery. International Journal of Molecular Sciences. 2019; 20(22):5661. https://doi.org/10.3390/ijms20225661
Chicago/Turabian StyleBeaurivage, Claudia, Elena Naumovska, Yee Xiang Chang, Edo D. Elstak, Arnaud Nicolas, Heidi Wouters, Guido van Moolenbroek, Henriëtte L. Lanz, Sebastiaan J. Trietsch, Jos Joore, and et al. 2019. "Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery" International Journal of Molecular Sciences 20, no. 22: 5661. https://doi.org/10.3390/ijms20225661
APA StyleBeaurivage, C., Naumovska, E., Chang, Y. X., Elstak, E. D., Nicolas, A., Wouters, H., van Moolenbroek, G., Lanz, H. L., Trietsch, S. J., Joore, J., Vulto, P., Janssen, R. A. J., Erdmann, K. S., Stallen, J., & Kurek, D. (2019). Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery. International Journal of Molecular Sciences, 20(22), 5661. https://doi.org/10.3390/ijms20225661