A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds



Abstract

1. Introduction
2. Results and Discussion
2.1. Shotgun Proteomics on the Test Protein BSA
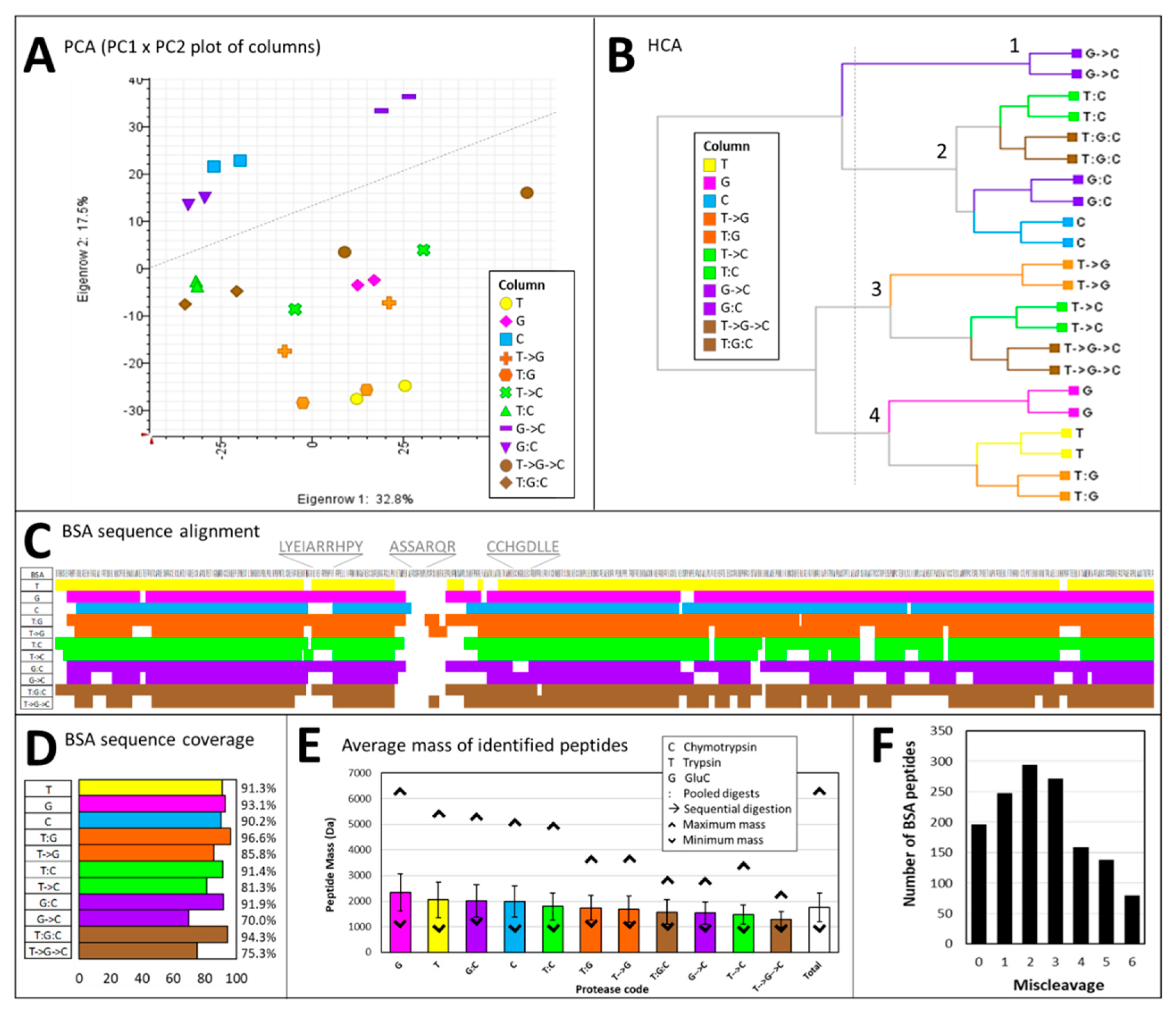
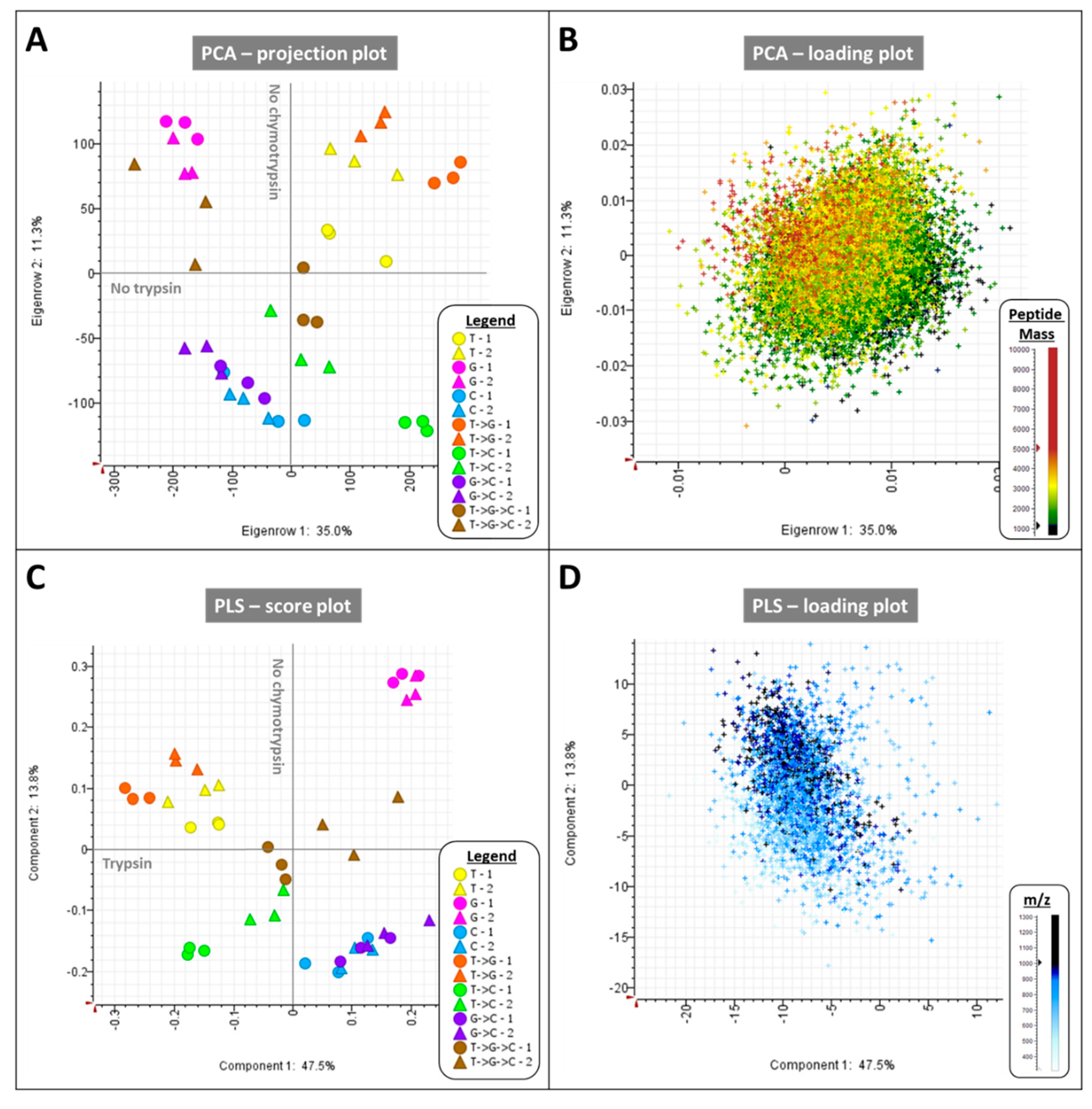
2.1.1. LC-MS/MS Data from BSA Digests Are Very Reproducible
2.1.2. Each Protease on Their Own or Combined Yield High Sequence Coverage of BSA
2.1.3. The Number of Miscleavages Is a Critical Parameter in Database Search
2.2. Medicinal Cannabis
2.2.1. LC-MS Patterns from Medicinal Cannabis Digests Are Very Complex
2.2.2. Sequential Enzymatic Digestions of Medicinal Cannabis Samples Augment the Success Rate of MS/MS Annotations in SEQUEST
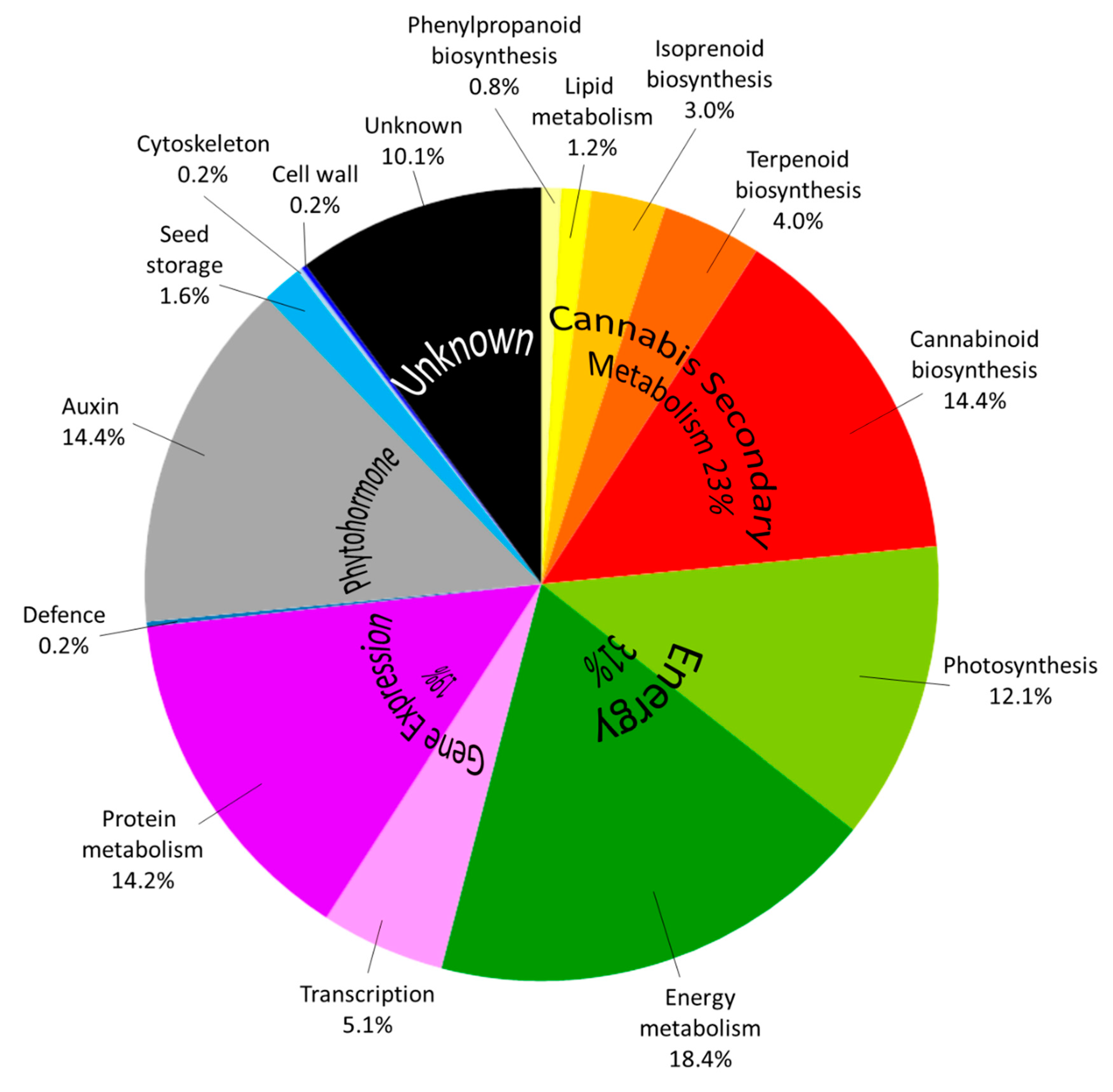
2.2.3. Proteins from Medicinal Cannabis Are Identified with High Sequence Coverage
3. Materials and Methods
3.1. Plant Materials and Chemicals
3.1.1. Apical Bud Sampling and Grinding
3.1.2. Chemicals
3.2. Protein Extraction Methods
3.3. Protein Assay
3.4. Protein Digestion
3.4.1. Digestion Using a Trypsin/LysC Protease Mix (T)
3.4.2. Digestion Using GluC (G)
3.4.3. Digestion Using Chymotrypsin (C)
3.4.4. Sequential Digestions Using Several Proteases (G->C, T->G, T->C, T->G->C)
3.4.5. Equimolar Mixtures of Digests (T:G, T:G, G:C, T:G:C)
3.5. Desalting
3.6. Peptide Digest Analysis by Nano Liquid Chromatography-Tandem Mass Spectrometry (nLC-MS/MS)
3.7. Database Search for Protein Identification and Annotation
3.8. Data Processing and Statistical Analyses
3.8.1. nLC-MS/MS Data Processing
3.8.2. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | amino acid |
| ACN | acetonitrile |
| BSA | bovine serum albumin |
| BUP | bottom-up proteomics |
| CBCA | cannabichromenic acid |
| CBDA | cannabidiolic acid |
| CBGA | cannabigerolic acid |
| FDR | false discovery rate |
| GluC | glutamyl peptidase I |
| GOT | geranylpyrophosphate:olivetolate geranyltransferase |
| HCA | hierarchical clustering analysis |
| kDa | kiloDalton |
| LysC | lysyl endopeptidase |
| MDP | middle-down proteomics |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| MW | molecular weight |
| nLC | nano-liquid chromatography |
| OAC | olivetolic acid cyclase |
| OLS | 3,5,7-trioxododecanoyl-CoA synthase |
| PCA | principal component analysis |
| PLS | partial least square |
| PTM | post-translational modification |
| RBCL | ribulose bisphosphate carboxylase large chain |
| SD | standard deviation |
| TDP | top-down proteomics |
| THCA | delta9-tetrahydrocannabinolicacid |
References
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, R. Chemistry and Analysis of Phytocannabinoids and Other Cannabis Constituents; Humana Press: New York, NY, USA, 2007. [Google Scholar]
- Fischedick, J.T.; Hazekamp, A.; Erkelens, T.; Choi, Y.H.; Verpoorte, R. Metabolic fingerprinting of Cannabis sativa l., cannabinoids and terpenoids for chemotaxonomic and drug standardization purposes. Phytochemistry 2010, 71, 2058–2073. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.M.; Elsohly, M.A.; Slade, D.; Ahmed, S.A.; Khan, I.A.; Ross, S.A. Biologically active cannabinoids from high-potency Cannabis sativa. J. Nat. Prod. 2009, 72, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol, a product isolated from the marihuana extract of minnesota wild hemp. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- Mechoulam, R.; Gaoni, Y. Recent advances in the chemistry of hashish. Prog. Chem. Org. Nat. Prod. 1967, 25, 175–213. [Google Scholar]
- Grassa, C.J.; Wenger, J.P.; Dabney, C.; Poplawski, S.G.; Motley, S.T.; Michael, T.P.; Schwartz, C.J.; Weiblen, G.D. A complete cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv 2018. [Google Scholar] [CrossRef]
- Laverty, K.U.; Stout, J.M.; Sullivan, M.J.; Shah, H.; Gill, N.; Holbrook, L.; Deikus, G.; Sebra, R.; Hughes, T.R.; Page, J.E.; et al. A physical and genetic map of Cannabis sativa identifies extensive rearrangement at the thc/cbd acid synthase locus. Genome Res. 2018, 29, 146–156. [Google Scholar] [CrossRef] [PubMed]
- van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Rochfort, S.; Spangenberg, G. Optimisation of protein extraction from medicinal cannabis mature buds for bottom-up proteomics. Molecules 2019, 24, 659. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Binos, S.; Rochfort, S.; Spangenberg, G. Top-down proteomics of medicinal cannabis. Proteomes 2019, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Aiello, G.; Fasoli, E.; Boschin, G.; Lammi, C.; Zanoni, C.; Citterio, A.; Arnoldi, A. Proteomic characterization of hempseed (Cannabis sativa L.). J. Proteom. 2016, 147, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Sergeant, K.; Leclercq, C.C.; Planchon, S.; Guignard, C.; Lenouvel, A.; Renaut, J.; Hausman, J.F.; Lutts, S.; Guerriero, G. Insights into the molecular regulation of monolignol-derived product biosynthesis in the growing hemp hypocotyl. BMC Plant Biol. 2018, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Bona, E.; Marsano, F.; Cavaletto, M.; Berta, G. Proteomic characterization of copper stress response in Cannabis sativa roots. Proteomics 2007, 7, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Happyana, N. Metabolomics, Proteomics, and Transcriptomics of Cannabis sativa L. Trichomes. Ph.D. Thesis, Bandung Institute of Technology, Bandung, West Java, Indonesia, 2014; pp. 1–212. [Google Scholar]
- Mamone, G.; Picariello, G.; Ramondo, A.; Nicolai, M.A.; Ferranti, P. Production, digestibility and allergenicity of hemp (Cannabis sativa L.) protein isolates. Food Res. Int. 2019, 115, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Seo, J.B.; Lee, M.Y. Proteomic profiling of hempseed proteins from cheungsam. Biochim. Biophys. Acta 2012, 1824, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Raharjo, T.J.; Widjaja, I.; Roytrakul, S.; Verpoorte, R. Comparative proteomics of Cannabis sativa plant tissues. J. Biomol. Tech. 2004, 15, 97–106. [Google Scholar] [PubMed]
- Rodziewicz, P.; Loroch, S.; Marczak, L.; Sickmann, A.; Kayser, O. Cannabinoid synthases and osmoprotective metabolites accumulate in the exudates of Cannabis sativa L. Glandular trichomes. Plant Sci. 2019, 284, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Hong, L.; Yang, Y.; Yanping, X.; Xing, H.; Gang, D. Protein changes in response to lead stress of lead-tolerant and lead-sensitive industrial hemp using swath technology. Genes 2019, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.K.; Vincent, D.; Zawadzki, J.L.; Savin, K.; Mertens, D.; Logan, A.; Williams, R.P.W. Investigation of age gelation in uht milk. Beverages 2018, 4, 95. [Google Scholar] [CrossRef]
- Vincent, D.; Elkins, A.; Condina, M.R.; Ezernieks, V.; Rochfort, S. Quantitation and identification of intact major milk proteins for high-throughput lc-esi-q-tof ms analyses. PLoS ONE 2016, 11, e0163471. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Ezernieks, V.; Elkins, A.; Nguyen, N.; Moate, P.J.; Cocks, B.G.; Rochfort, S. Milk bottom-up proteomics: Method optimization. Front. Genet. 2015, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Mertens, D.; Rochfort, S. Optimisation of milk protein top-down sequencing using in-source collision-induced dissociation in the maxis quadrupole time-of-flight mass spectrometer. Molecules 2018, 23, 2777. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Papasotiriou, D.G.; Karas, M. 100% protein sequence coverage: A modern form of surrealism in proteomics. Amino Acids 2011, 41, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Switzar, L.; Giera, M.; Niessen, W.M.A. Protein digestion: An overview of the available techniques and recents developments. J. Proteome Res. 2013, 12, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef] [PubMed]
- Vandermarliere, E.; Mueller, M.; Martens, L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 2013, 32, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Ong, S.E.; Mann, M. Trypsin cleaves exclusively c-terminal to arginine and lysine residues. Mol. Cell Proteom. 2004, 3, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Gershon, P.D. Cleaved and missed sites for trypsin, lys-c, and lys-n can be predicted with high confidence on the basis of sequence context. J. Proteome Res. 2014, 13, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Slechtova, T.; Gilar, M.; Kalikova, K.; Tesarova, E. Insight into trypsin miscleavage: Comparison of kinetic constants of problematic peptide sequences. Anal. Chem. 2015, 87, 7636–7643. [Google Scholar] [CrossRef] [PubMed]
- Glatter, T.; Ludwig, C.; Ahrne, E.; Aebersold, R.; Heck, A.J.; Schmidt, A. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem lys-c/trypsin proteolysis over trypsin digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Klammer, A.A.; MacCoss, M.J. Effects of modified digestion schemes on the identification of proteins from complex mixtures. J. Proteome Res. 2006, 5, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R. Quantitative evaluation of filter aided sample preparation (FASP) and multienzyme digestion fasp protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, G.R.; Boily, Y.; Houmard, J. Purification and properties of an extracellular protease of staphylococcus aureus. J. Biol. Chem. 1972, 247, 6720–6726. [Google Scholar] [PubMed]
- Cristobal, A.; Marino, F.; Post, H.; van den Toorn, H.W.; Mohammed, S.; Heck, A.J. Toward an optimized workflow for middle-down proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [PubMed]
- Moradian, A.; Kalli, A.; Sweredoski, M.J.; Hess, S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 2014, 14, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Garcia, B.A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteom. 2017, 14, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.V.; Wang, T.; Young, N.L. One-pot quantitative top- and middle-down analysis of gluc-digested histone h4. J. Am. Soc. Mass Spectrom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.M.; Zang, L.; Sebastiano, R.; Santos, M.R.; Bush, D.R.; Karger, B.L.; Ivanov, A.R. Complementary middle-down and intact monoclonal antibody proteoform characterization by capillary zone electrophoresis - mass spectrometry. Electrophoresis 2018, 39, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- MacCoss, M.J.; McDonald, W.H.; Saraf, A.; Sadygov, R.; Clark, J.M.; Tasto, J.J.; Gould, K.L.; Wolters, D.; Washburn, M.; Weiss, A.; et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. USA 2002, 99, 7900–7905. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, A.; Vanselow, J.T.; Kramer, A. Mapping of phosphorylation sites by a multi-protease approach with specific phosphopeptide enrichment and nanolc-ms/ms analysis. Anal. Chem. 2005, 77, 5243–5250. [Google Scholar] [CrossRef] [PubMed]
- Trevisan-Silva, D.; Bednaski, A.V.; Fischer, J.S.G.; Veiga, S.S.; Bandeira, N.; Guthals, A.; Marchini, F.K.; Leprevost, F.V.; Barbosa, V.C.; Senff-Ribeiro, A.; et al. A multi-protease, multi-dissociation, bottom-up-to-top-down proteomic view of the loxosceles intermedia venom. Sci. Data 2017, 4, 170090. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Poetsch, A. Protein cleavage strategies for an improved analysis of the membrane proteome. Proteome Sci. 2006, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Wolters, D.; Rogner, M.; Poetsch, A. Toward the complete membrane proteome: High coverage of integral membrane proteins through transmembrane peptide detection. Mol. Cell Proteom. 2006, 5, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Less is more: Membrane protein digestion beyond urea-trypsin solution for next-level proteomics. Mol. Cell Proteom. 2015, 14, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Biringer, R.G.; Amato, H.; Harrington, M.G.; Fonteh, A.N.; Riggins, J.N.; Hühmer, A.F. Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap lc-ms/ms. Brief Funct. Genom. Proteom. 2006, 5, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Wu, S.L.; Shieh, P.; Hancock, W.S. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary lc with ion trap ms/ms. J. Proteome Res. 2003, 2, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Trudgian, D.C.; Lemoff, A.; Yadavalli, S.; Mirzaei, H. Confetti: A multiprotease map of the hela proteome for comprehensive proteomics. Mol. Cell Proteom. 2014, 13, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, D.D.; Silva, A.M.; Vitorino, R.; Domingues, M.R.; Domingues, P. The efficiency of trypsin digestion for mass-spectrometry-based identification and quantification of oxidized proteins: Evaluation of the digestion of oxidized bovine serum albumin. Eur. J. Mass Spectrom. 2014, 20, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.H.; Liu, C.C.; Chen, B.; Yan, L.; Yu, H.L.; Yang, Y.; Wu, J.N.; Li, X.S.; Liu, S.L. Lc-hrms screening and identification of novel peptide markers of ricin based on multiple protease digestion strategies. Toxins 2019, 11, 393. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.S. Protein identification using ms/ms data. J. Proteom. 2011, 74, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Stead, D.A.; Preece, A.; Brown, A.J. Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol. Cell Proteom. 2006, 5, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the ms/ms spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed]
- Giannone, R.J.; Wurch, L.L.; Podar, M.; Hettich, R.L. Rescuing those left behind: Recovering and characterizing underdigested membrane and hydrophobic proteins to enhance proteome measurement depth. Anal. Chem. 2015, 87, 7720–7728. [Google Scholar] [CrossRef] [PubMed]
- Vermachova, M.; Purkrtova, Z.; Santrucek, J.; Jolivet, P.; Chardot, T.; Kodicek, M. New protein isoforms identified within arabidopsis thaliana seed oil bodies combining chymotrypsin/trypsin digestion and peptide fragmentation analysis. Proteomics 2011, 11, 3430–3434. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M. Cannabis systematics at the levels of family, genus, and species. Cannabis Cannabinoid. Res. 2018, 3, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Page, J.; Boubakir, Z. Aromatic Prenyltransferase from Cannabis. WO/2011/017798, 17 February 2011. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tube | 1. MS | 2. all MS/MS | % MS/MS | 3. SEQUEST Annotated MS/MS | % MS/MS Annotated | % MS Annotated | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Protease Mix | Rep 1 | Rep 2 | Mean | SD | % CV | Rep 1 | Rep 2 | Mean | SD | Percent | Rep 1 | Rep 2 | Mean | SD | % | % |
| BSA | T | 83,678 | 83,056 | 83,367 | 440 | 0.5 | 9769 | 9325 | 9547 | 314 | 11 | 2133 | 1875 | 2004 | 182 | 21 | 2.4 |
| BSA | G | 91,922 | 98,895 | 95,409 | 3487 | 3.7 | 9081 | 9628 | 9355 | 387 | 10 | 929 | 1363 | 1146 | 307 | 12 | 1.2 |
| BSA | C | 92,116 | 90,303 | 91,210 | 907 | 1.0 | 10,327 | 9792 | 10,060 | 378 | 11 | 1358 | 1267 | 1313 | 64 | 13 | 1.4 |
| BSA | T->G | 89,648 | 83,107 | 86,378 | 3271 | 3.8 | 11,311 | 9698 | 10,505 | 1141 | 12 | 2178 | 1978 | 2078 | 141 | 20 | 2.4 |
| BSA | T:G | 84,347 | 87,462 | 85,905 | 1558 | 1.8 | 8605 | 9720 | 9163 | 788 | 11 | 2141 | 2332 | 2237 | 135 | 24 | 2.6 |
| BSA | T->C | 87,203 | 79,616 | 83,410 | 3794 | 4.5 | 10,944 | 8810 | 9877 | 1509 | 12 | 1864 | 1549 | 1707 | 223 | 17 | 2.0 |
| BSA | T:C | 90,847 | 92,736 | 91,792 | 945 | 1.0 | 10,245 | 10,115 | 10,180 | 92 | 11 | 2428 | 1931 | 2180 | 351 | 21 | 2.4 |
| BSA | G->C | 77,085 | 82,055 | 79,570 | 2485 | 3.1 | 6450 | 5163 | 5807 | 910 | 7 | 1103 | 475 | 789 | 444 | 14 | 1.0 |
| BSA | G:C | 99,001 | 100,001 | 99,501 | 500 | 0.5 | 9980 | 9847 | 9914 | 94 | 10 | 1169 | 1065 | 1117 | 74 | 11 | 1.1 |
| BSA | T->G->C | 88,919 | 84,798 | 86,859 | 2061 | 2.4 | 9880 | 6137 | 8009 | 2647 | 9 | 1485 | 1005 | 1245 | 339 | 16 | 1.4 |
| BSA | T:G:C | 91,975 | 89,420 | 90,698 | 1278 | 1.4 | 10,201 | 9503 | 9852 | 494 | 11 | 1015 | 1616 | 1316 | 425 | 13 | 1.5 |
| BSA | mean | 88,795 | 88,314 | 88,554 | 1884 | 2 | 9708 | 8885 | 9297 | 796 | 10 | 1618 | 1496 | 1557 | 244 | 17 | 2 |
| BSA | SD | 5707 | 6752 | 5811 | 1218 | 1 | 1317 | 1648 | 1333 | 756 | 1 | 544 | 531 | 501 | 136 | 4 | 1 |
| min | 77,085 | 79,616 | 79,570 | 440 | 1 | 6450 | 5163 | 5807 | 92 | 7 | 929 | 475 | 789 | 64 | 11 | 1 | |
| max | 99,001 | 100,001 | 99,501 | 3794 | 5 | 11,311 | 10,115 | 10,505 | 2647 | 12 | 2428 | 2332 | 2237 | 444 | 24 | 3 | |
| Tube | 1. MS | 2. all MS/MS | % MS/MS | 3. SEQUEST Annotated MS/MS | % MS/MS Annotated | % MS Annotated | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biol rep | Protease mix | Rep 1 | Rep 2 | Mean | SD | % CV | Rep 1 | Rep 2 | Mean | SD | Percent | Rep 1 | Rep 2 | Mean | SD | % | % | |
| Bud 1 | T | 86,458 | 115,577 | 101,018 | 20,590 | 20.4 | 12,827 | 11,731 | 12,279 | 775 | 12 | 2042 | 1929 | 1986 | 80 | 16 | 2.0 | |
| Bud 2 | T | 72,907 | 113,303 | 93,105 | 28,564 | 30.7 | 10,775 | 11,160 | 10,968 | 272 | 12 | 1606 | 1740 | 1673 | 95 | 15 | 1.8 | |
| Bud 3 | T | 70,473 | 112,818 | 91,646 | 29,942 | 32.7 | 10,541 | 10,585 | 10,563 | 31 | 12 | 1513 | 1643 | 1578 | 92 | 15 | 1.7 | |
| Bud 1 | G | 106,622 | 84,761 | 95,692 | 15,458 | 16.2 | 9035 | 8501 | 8768 | 378 | 9 | 1388 | 1376 | 1382 | 8 | 16 | 1.4 | |
| Bud 2 | G | 95,761 | 88,387 | 92,074 | 5214 | 5.7 | 8032 | 7906 | 7969 | 89 | 9 | 1200 | 1146 | 1173 | 38 | 15 | 1.3 | |
| Bud 3 | G | 93,760 | 91,846 | 92,803 | 1353 | 1.5 | 8810 | 8115 | 8463 | 491 | 9 | 1326 | 1290 | 1308 | 25 | 15 | 1.4 | |
| Bud 1 | C | 93,117 | 95,399 | 94,258 | 1614 | 1.7 | 9486 | 8644 | 9065 | 595 | 10 | 2589 | 2200 | 2395 | 275 | 26 | 2.5 | |
| Bud 2 | C | 93,778 | 92,536 | 93,157 | 878 | 0.9 | 8433 | 7788 | 8111 | 456 | 9 | 2232 | 1857 | 2045 | 265 | 25 | 2.2 | |
| Bud 3 | C | 97,359 | 97,813 | 97,586 | 321 | 0.3 | 9508 | 8341 | 8925 | 825 | 9 | 2382 | 2098 | 2240 | 201 | 25 | 2.3 | |
| Bud 1 | T->G | 116,131 | 113,352 | 114,742 | 1965 | 1.7 | 11,909 | 11,406 | 11,658 | 356 | 10 | 3416 | 3163 | 3290 | 179 | 28 | 2.9 | |
| Bud 2 | T->G | 113,690 | 111,601 | 112,646 | 1477 | 1.3 | 11,511 | 10,857 | 11,184 | 462 | 10 | 3103 | 2904 | 3004 | 141 | 27 | 2.7 | |
| Bud 3 | T->G | 118,020 | 115,958 | 116,989 | 1458 | 1.2 | 12,362 | 11,811 | 12,087 | 390 | 10 | 3633 | 3405 | 3519 | 161 | 29 | 3.0 | |
| Bud 1 | T->C | 98,125 | 94,395 | 96,260 | 2638 | 2.7 | 10,963 | 9568 | 10,266 | 986 | 11 | 4066 | 3434 | 3750 | 447 | 37 | 3.9 | |
| Bud 2 | T->C | 98,455 | 97,615 | 98,035 | 594 | 0.6 | 10,622 | 9090 | 9856 | 1083 | 10 | 4024 | 3308 | 3666 | 506 | 37 | 3.7 | |
| Bud 3 | T->C | 100,667 | 97,679 | 99,173 | 2113 | 2.1 | 11,238 | 8873 | 10,056 | 1672 | 10 | 4297 | 3321 | 3809 | 690 | 38 | 3.8 | |
| Bud 1 | G->C | 92,277 | 90,930 | 91,604 | 952 | 1.0 | 8219 | 7625 | 7922 | 420 | 9 | 2786 | 2545 | 2666 | 170 | 34 | 2.9 | |
| Bud 2 | G->C | 86,056 | 83,949 | 85,003 | 1490 | 1.8 | 7160 | 6390 | 6775 | 544 | 8 | 2393 | 2190 | 2292 | 144 | 34 | 2.7 | |
| Bud 3 | G->C | 93,847 | 89,624 | 91,736 | 2986 | 3.3 | 8158 | 7398 | 7778 | 537 | 8 | 2687 | 2502 | 2595 | 131 | 33 | 2.8 | |
| Bud 1 | T->G->C | 88,886 | 56,861 | 72,874 | 22,645 | 31.1 | 9479 | 4279 | 6879 | 3677 | 9 | 4117 | 2002 | 3060 | 1496 | 44 | 4.2 | |
| Bud 2 | T->G->C | 67,123 | 49,316 | 58,220 | 12,591 | 21.6 | 6835 | 1770 | 4303 | 3581 | 7 | 3065 | 824 | 1945 | 1585 | 45 | 3.3 | |
| Bud 3 | T->G->C | 84,077 | 77,062 | 80,570 | 4960 | 6.2 | 7685 | 5570 | 6628 | 1496 | 8 | 3392 | 2524 | 2958 | 614 | 45 | 3.7 | |
| Mean | 13,559 | 17,773 | 13,095 | 9797 | 11 | 1743 | 2526 | 2047 | 992 | 1 | 991 | 787 | 836 | 439 | 10 | 1 | ||
| SD | 13,232 | 17,345 | 12,779 | 9561 | 11 | 1701 | 2465 | 1997 | 968 | 1 | 967 | 769 | 816 | 428 | 10 | 1 | ||
| Min | 67,123 | 49,316 | 58,220 | 321 | 0.33 | 6835 | 1770 | 4303 | 31.1 | 7.391 | 1200 | 824 | 1173 | 8.49 | 14.7195 | 1.27398 | ||
| Max | 118,020 | 115,958 | 116,989 | 29,942 | 32.7 | 12,827 | 11,811 | 12,279 | 3677 | 12.155 | 4297 | 3434 | 3809 | 1585 | 45.1894 | 4.19837 | ||
| Proteases | Carbamidomethylation | Acetylation | Phosphorylation | Oxidation | Total |
|---|---|---|---|---|---|
| Trypsin/LysC | 1362 | 296 | 6213 | 2927 | 10,798 |
| Chymotrypsin | 1483 | 238 | 7683 | 3520 | 12,924 |
| GluC | 1396 | 149 | 4820 | 2789 | 9154 |
| Total | 4241 | 683 | 18,716 | 9236 | 32,876 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Ezernieks, V.; Rochfort, S.; Spangenberg, G. A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds. Int. J. Mol. Sci. 2019, 20, 5630. https://doi.org/10.3390/ijms20225630
Vincent D, Ezernieks V, Rochfort S, Spangenberg G. A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds. International Journal of Molecular Sciences. 2019; 20(22):5630. https://doi.org/10.3390/ijms20225630
Chicago/Turabian StyleVincent, Delphine, Vilnis Ezernieks, Simone Rochfort, and German Spangenberg. 2019. "A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds" International Journal of Molecular Sciences 20, no. 22: 5630. https://doi.org/10.3390/ijms20225630
APA StyleVincent, D., Ezernieks, V., Rochfort, S., & Spangenberg, G. (2019). A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds. International Journal of Molecular Sciences, 20(22), 5630. https://doi.org/10.3390/ijms20225630