The Rich World of p53 DNA Binding Targets: The Role of DNA Structure



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Various DNA Targets of p53
2.1. ChIP-Seq and p53-Target Sequences
2.2. p53 Binding to Distorted Double-Stranded DNA
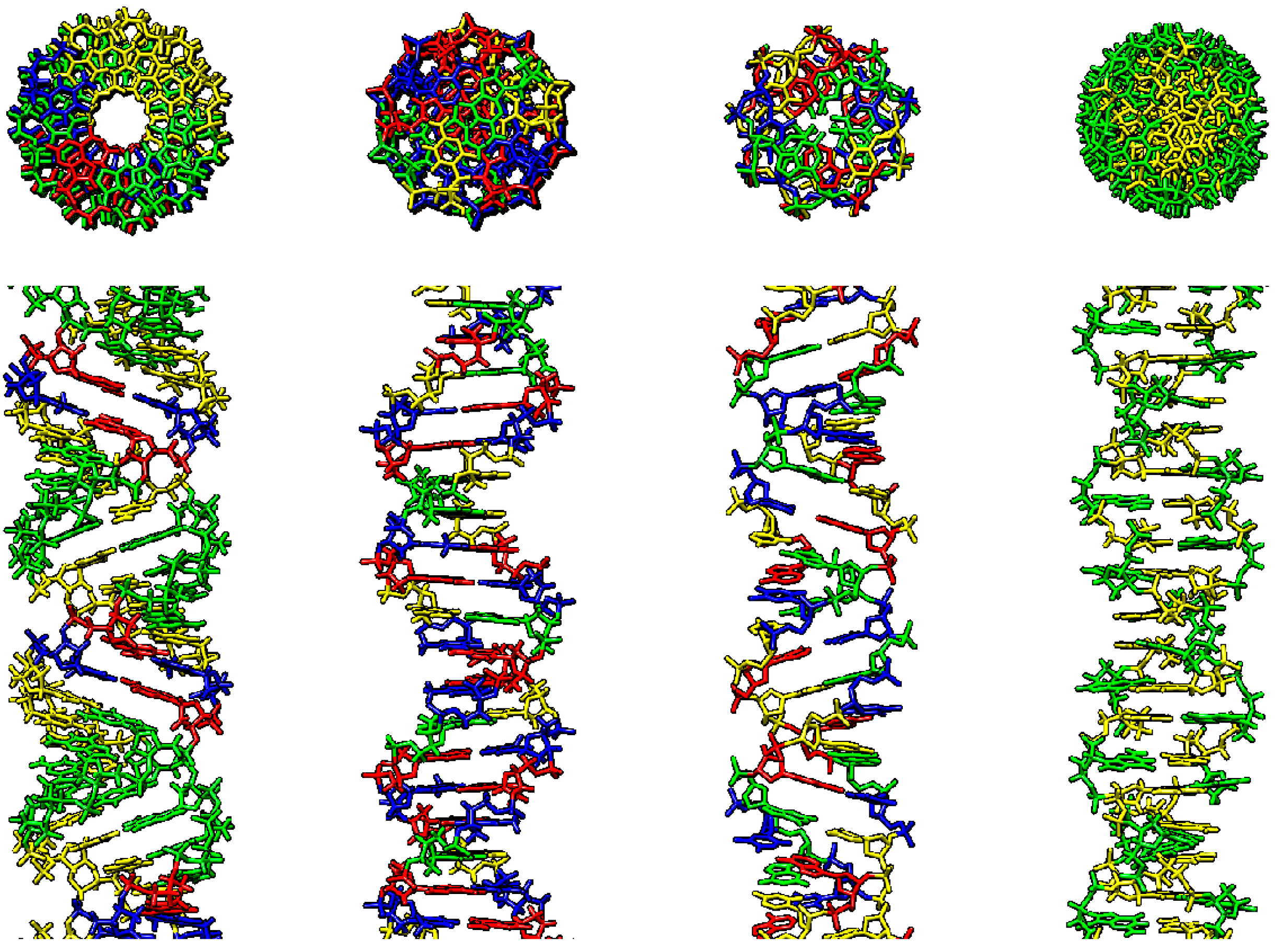
2.3. p53 Binding to DNA Structures Presented in Single-, Triple- and Four-Stranded DNA Motifs
2.3.1. Hemicatenate DNA
2.3.2. Telomeric T-Loops
2.3.3. Three-Stranded Structure
2.3.4. Four-Stranded Structures
2.3.5. Loops and Cruciforms
2.3.6. Influence of Epigenetic Changes to p53-DNA Interactions
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Gohler, T.; Reimann, M.; Cherny, D.; Walter, K.; Warnecke, G.; Kim, E.; Deppert, W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J. Biol. Chem. 2002, 277, 41192–41203. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, H.E.; Bodey, B. The role of apoptosis in normal ontogenesis and solid human neoplasms. In Vivo 2000, 14, 789–803. [Google Scholar] [PubMed]
- Kylarová, D.; Vrchovecký, J.; Holinka, M.; Erdösová, B. The occurrence of c-myc, p53 and Bcl-2 family proteins in the early phase of development of duodenal epithelium. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2004, 148, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, J.; Lichnovsky, V.; Kylarova, D.; Erdosova, B.; Vranka, P. Involvement of p53 and Bcl-2 family proteins in regulating programmed cell death and proliferation in human embryogenesis. Gen. Physiol. Biophys. 2004, 23, 209–229. [Google Scholar]
- Porrello, A.; Cerone, M.A.; Coen, S.; Gurtner, A.; Fontemaggi, G.; Cimino, L.; Piaggio, G.; Sacchi, A.; Soddu, S. P53 Regulates Myogenesis by Triggering the Differentiation Activity of pRb. J. Cell Biol. 2000, 151, 1295–1304. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Kaiser, A.M.; Attardi, L.D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Iwakuma, T. Non-Canonical Cell Death Induced by p53. Int. J. Mol. Sci. 2016, 17, 2068. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, M.J.; Mukhopadhyay, S.; Hoofnagle, M.; Chabasse, C.; Sarkar, R. Tumor suppressor protein p53 negatively regulates ischemia-induced angiogenesis and arteriogenesis. J. Vasc. Surg. 2018, 68, 222S–233S.e1. [Google Scholar] [CrossRef] [PubMed]
- Chandrangsu, S.; Sappayatosok, K. p53, p63 and p73 expression and angiogenesis in keratocystic odontogenic tumors. J. Clin. Exp. Dent. 2016, 8, e505–e511. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, S.; Rossi, A.; Di Daniele, N.; Melino, G.; Annicchiarico-Petruzzelli, M.; Raschellà, G. DNA repair and aging: The impact of the p53 family. Aging 2015, 7, 1050–1065. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013. [Google Scholar] [CrossRef]
- Hussain, M.; Tian, K.; Mutti, L.; Krstic-Demonacos, M.; Schwartz, J.-M. The Expanded p53 Interactome as a Predictive Model for Cancer Therapy. Genom. Comput. Biol. 2015, 1, e20. [Google Scholar] [CrossRef][Green Version]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Valenti, F.; Fausti, F.; Biagioni, F.; Shay, T.; Fontemaggi, G.; Domany, E.; Yaffe, M.B.; Strano, S.; Blandino, G.; Di Agostino, S. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 2011, 10, 4330–4340. [Google Scholar] [CrossRef]
- Liu, K.; Ling, S.; Lin, W.-C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [PubMed]
- Stindt, M.H.; Muller, P.A.J.; Ludwig, R.L.; Kehrloesser, S.; Dötsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A.; Frazier, D.P. Transcription factors that interact with p53 and Mdm2. Int. J. Cancer 2016, 138, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Woods, Y.L.; Lane, D.P. Exploiting the p53 pathway for cancer diagnosis and therapy. Hematol. J. 2003, 4, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Lane, D.P. Exploiting the p53 Pathway for Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026310. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Lohrum, M.A.E.; Vousden, K.H. Regulation and activation of p53 and its family members. Cell Death Differ. 1999, 6, 1162–1168. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Du, J. Cellular adaptation to hypoxia and p53 transcription regulation. J. Zhejiang Univ.-Sci. B 2009, 10, 404–410. [Google Scholar] [CrossRef]
- Itoh, Y.; Murata, A.; Sakamoto, S.; Nanatani, K.; Wada, T.; Takahashi, S.; Kamagata, K. Activation of p53 Facilitates the Target Search in DNA by Enhancing the Target Recognition Probability. J. Mol. Biol. 2016, 428, 2916–2930. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Timsit, Y. DNA Self-Assembly: From Chirality to Evolution. Int. J. Mol. Sci. 2013, 14, 8252–8270. [Google Scholar] [CrossRef] [PubMed]
- Frees, S.; Menendez, C.; Crum, M.; Bagga, P.S. QGRS-Conserve: A computational method for discovering evolutionarily conserved G-quadruplex motifs. Hum. Genom. 2014, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E. Local supercoil-stabilized DNA structures. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 151–226. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.; Muskhelishvili, G. DNA structure and function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef]
- Yahyaoui, W.; Callejo, M.; Price, G.B.; Zannis-Hadjopoulos, M. Deletion of the cruciform binding domain in CBP/14-3-3 displays reduced origin binding and initiation of DNA replication in budding yeast. BMC Mol. Biol. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef]
- Brazdova, M.; Tichy, V.; Helma, R.; Bazantova, P.; Polaskova, A.; Krejci, A.; Petr, M.; Navratilova, L.; Ticha, O.; Nejedly, K.; et al. p53 Specifically Binds Triplex DNA In Vitro and in Cells. PLoS ONE 2016, 11, e0167439. [Google Scholar] [CrossRef]
- Waller, Z.A.; Pinchbeck, B.J.; Buguth, B.S.; Meadows, T.G.; Richardson, D.J.; Gates, A.J. Control of bacterial nitrate assimilation by stabilization of G-quadruplex DNA. Chem. Commun. 2016, 52, 13511–13514. [Google Scholar] [CrossRef]
- Bartas, M.; Čutová, M.; Brázda, V.; Kaura, P.; Št’astnỳ, J.; Kolomazník, J.; Coufal, J.; Goswami, P.; Červeň, J.; Pečinka, P. The Presence and Localization of G-Quadruplex Forming Sequences in the Domain of Bacteria. Molecules 2019, 24, 1711. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2006, 35, 406–413. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Rajeswari, M.R. DNA triplex structures in neurodegenerative disorder, Friedreich’s ataxia. J. Biosci. 2012, 37, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Helma, R.; Bažantová, P.; Petr, M.; Adámik, M.; Renčiuk, D.; Tichý, V.; Pastuchová, A.; Soldánová, Z.; Pečinka, P.; Bowater, R.P.; et al. p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia. Molecules 2019, 24, 2078. [Google Scholar] [CrossRef] [PubMed]
- Cimino-Reale, G.; Zaffaroni, N.; Folini, M. Emerging Role of G-quadruplex DNA as Target in Anticancer Therapy. Curr. Pharm. Des. 2016, 22, 6612–6624. [Google Scholar] [CrossRef]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Pan, Y.; Zheng, J.; Levine, A.J.; Nussinov, R. Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic Acids Res. 2007, 35, 2986–3001. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.T.; Weng, Z.P.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. Potentiating the p53 network. Discov. Med. 2010, 10, 94–100. [Google Scholar]
- Wang, B.; Xiao, Z.; Ren, E.C. Redefining the p53 response element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elite 2014, 3, e02200. [Google Scholar]
- Chang, G.S.; Chen, X.A.; Park, B.; Rhee, H.S.; Li, P.; Han, K.H.; Mishra, T.; Chan-Salis, K.Y.; Li, Y.; Hardison, R.C.; et al. A Comprehensive and High-Resolution Genome-wide Response of p53 to Stress. Cell Rep. 2014, 8, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.-H.; Chao, C.-F.; Huang, H.-C.; Lee, H.-Y.; Kannagi, R.; Chen, J.-Y. Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. Int. J. Mol. Sci. 2019, 20, 3681. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45. [Google Scholar] [CrossRef]
- Qian, H.; Wang, T.; Naumovski, L.; Lopez, C.D.; Brachmann, R.K. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene 2002, 21, 7901–7911. [Google Scholar] [CrossRef]
- Veprintsev, D.B.; Fersht, A.R. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008, 36, 1589–1598. [Google Scholar] [CrossRef]
- Brazda, V.; Kolomaznik, J.; Lysek, J.; Haronikova, L.; Coufal, J.; Stastny, J. Palindrome analyser—A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem. Biophys. Res. Commun. 2016, 478, 1739–1745. [Google Scholar] [CrossRef]
- McKinney, K.; Mattia, M.; Gottifredi, V.; Prives, C. p53 linear diffusion along DNA requires its C terminus. Mol. Cell 2004, 16, 413–424. [Google Scholar] [CrossRef]
- Brazda, V.; Jagelska, E.B.; Fojta, M.; Palecek, E. Searching for target sequences by p53 protein is influenced by DNA length. Biochem. Biophys. Res. Commun. 2006, 341, 470–477. [Google Scholar] [CrossRef]
- Itoh, Y.; Murata, A.; Takahashi, S.; Kamagata, K. Intrinsically disordered domain of tumor suppressor p53 facilitates target search by ultrafast transfer between different DNA strands. Nucleic Acids Res. 2018, 46, 7261–7269. [Google Scholar] [CrossRef]
- Nguyen, T.-A.T.; Grimm, S.A.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; Lavender, C.A.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a human p53 universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Raney, B.J.; Dreszer, T.R.; Barber, G.P.; Clawson, H.; Fujita, P.A.; Wang, T.; Nguyen, N.; Paten, B.; Zweig, A.S.; Karolchik, D.; et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015, 16, 464. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wu, H.; Zhu, X.; Guo, X.; Hutchins, A.P.; Luo, Z.; Song, H.; Chen, Y.; Lai, K.; Yin, M.; et al. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res. 2015, 25, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Pivonkova, H.; Brazdova, M.; Kasparkova, J.; Brabec, V.; Fojta, M. Recognition of cisplatin-damaged DNA by p53 protein: Critical role of the p53 C-terminal domain. Biochem. Biophys. Res. Commun. 2006, 339, 477–484. [Google Scholar] [CrossRef]
- Degtyareva, N.; Subramanian, D.; Griffith, J.D. Analysis of the binding of p53 to DNAs containing mismatched and bulged bases. J. Biol. Chem. 2001, 276, 8778–8784. [Google Scholar] [CrossRef]
- Walter, K.; Warnecke, G.; Bowater, R.; Deppert, W.; Ella, K. Tumor suppressor p53 binds with high affinity to CTG center dot CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. J. Biol. Chem. 2005, 280, 42497–42507. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Chai, X.; Johnston, K.; Clements, A.; Marmorstein, R. Crystal structure of the mouse p53 core DNA-binding domain at 2.7 A resolution. J. Biol. Chem. 2001, 276, 12120–12127. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Allen, M.D.; Fersht, A.R. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef]
- Suad, O.; Rozenberg, H.; Brosh, R.; Diskin-Posner, Y.; Kessler, N.; Shimon, L.J.W.; Frolow, F.; Liran, A.; Rotter, V.; Shakked, Z. Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations. J. Mol. Biol. 2009, 385, 249–265. [Google Scholar] [CrossRef]
- Malecka, K.A.; Ho, W.C.; Marmorstein, R. Crystal structure of a p53 core tetramer bound to DNA. Oncogene 2009, 28, 325–333. [Google Scholar] [CrossRef][Green Version]
- Nagaich, A.K.; Appella, E.; Harrington, R.E. DNA bending is essential for the site-specific recognition of DNA response elements by the DNA binding domain of the tumor suppressor protein p53. J. Biol. Chem. 1997, 272, 14842–14849. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Bakalkin, G.; Yakovleva, T.; Selivanova, G.; Magnusson, K.P.; Szekely, L.; Kiseleva, E.; Klein, G.; Terenius, L.; Wiman, K.G. p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc. Natl. Acad. Sci. USA 1994, 91, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Bakalkin, G.; Selivanova, G.; Yakovleva, T.; Kiseleva, E.; Kashuba, E.; Magnusson, K.P.; Szekely, L.; Klein, G.; Terenius, L.; Wiman, K.G. p53 binds single-stranded DNA ends through the C-terminal domain and internal DNA segments via the middle domain. Nucleic Acids Res. 1995, 23, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Coufal, J. Recognition of local DNA structures by p53 protein. Int. J. Mol. Sci. 2017, 18, 375. [Google Scholar] [CrossRef] [PubMed]
- Palecek, E.; Vlk, D.; Stankova, V.; Brazda, V.; Vojtesek, B.; Hupp, T.R.; Schaper, A.; Jovin, T.M. Tumor suppressor protein p53 binds preferentially to supercoiled DNA. Oncogene 1997, 15, 2201–2209. [Google Scholar] [CrossRef]
- Palecek, E.; Brazda, V.; Jagelska, E.; Pecinka, P.; Karlovska, L.; Brazdova, M. Enhancement of p53 sequence-specific binding by DNA supercoiling. Oncogene 2004, 23, 2119–2127. [Google Scholar] [CrossRef]
- Brázda, V.; Paleĉek, J.; Pospísilová, S.; Vojtêsek, B.; Paleĉek, E. Specific modulation of p53 binding to consensus sequence within supercoiled DNA by monoclonal antibodies. Biochem. Biophys. Res. Commun. 2000, 267, 934–939. [Google Scholar] [CrossRef]
- Palecek, E.; Brazdova, M.; Brazda, V.; Palecek, J.; Billova, S.; Subramaniam, V.; Jovin, T.M. Binding of p53 and its core domain to supercoiled DNA. Eur. J. Biochem. 2001, 268, 573–581. [Google Scholar] [CrossRef]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential Binding of Hot Spot Mutant p53 Proteins to Supercoiled DNA In Vitro and in Cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef]
- Mazur, S.J.; Sakaguchi, K.; Appella, E.; Wang, X.W.; Harris, C.C.; Bohr, V.A. Preferential binding of tumor suppressor p53 to positively or negatively supercoiled DNA involves the C-terminal domain. J. Mol. Biol. 1999, 292, 241–249. [Google Scholar] [CrossRef]
- Pivonkova, H.; Sebest, P.; Pecinka, P.; Ticha, O.; Nemcova, K.; Brazdova, M.; Jagelska, E.B.; Brazda, V.; Fojta, M. Selective binding of tumor suppressor p53 protein to topologically constrained DNA: Modulation by intercalative drugs. Biochem. Biophys. Res. Commun. 2010, 393, 894–899. [Google Scholar] [CrossRef]
- Sandri, M.I.; Isaacs, R.J.; Ongkeko, W.M.; Harris, A.L.; Hickson, I.D.; Broggini, M.; Vikhanskaya, F. p53 regulates the minimal promoter of the human topoisomerase IIalpha gene. Nucleic Acids Res. 1996, 24, 4464–4470. [Google Scholar] [CrossRef] [PubMed]
- Jagelska, E.B.; Brazda, V.; Pecinka, P.; Palecek, E.; Fojta, M. DNA topology influences p53 sequence-specific DNA binding through structural transitions within the target sites. Biochem. J. 2008, 412, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Zhang, F.; Lupski, J.R. Structural variation of the human genome: Mechanisms, assays, and role in male infertility. Syst. Biol. Reprod. Mech. 2011, 57, 3–16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Warburton, P.E.; Giordano, J.; Cheung, F.; Gelfand, Y.; Benson, G. Inverted repeat structure of the human genome: The X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004, 14, 1861–1869. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef]
- Lee, S.; Ho, J.Y.; Liu, J.J.; Lee, H.; Park, J.Y.; Baik, M.; Ko, M.; Lee, S.U.; Choi, Y.J.; Hur, S.Y. CKD-602, a topoisomerase I inhibitor, induces apoptosis and cell-cycle arrest and inhibits invasion in cervical cancer. Mol. Med. 2019, 25, 23. [Google Scholar] [CrossRef]
- Zheng, K.-W.; He, Y.; Liu, H.-H.; Li, X.-M.; Hao, Y.-H.; Tan, Z. Superhelicity Constrains a Localized and R-Loop-Dependent Formation of G-Quadruplexes at the Upstream Region of Transcription. ACS Chem. Biol. 2017, 12, 2609–2618. [Google Scholar] [CrossRef]
- Lee, S.M.; Cavallo, L.; Griffith, J. Human p53 binds Holliday junctions strongly and facilitates their cleavage. J. Biol. Chem. 1997, 272, 7532–7539. [Google Scholar] [CrossRef]
- Subramanian, D.; Griffith, J.D. p53 Monitors replication fork regression by binding to “chickenfoot” intermediates. J. Biol. Chem. 2005, 280, 42568–42572. [Google Scholar] [CrossRef]
- Stansel, R.M.; Subramanian, D.; Griffith, J.D. p53 binds telomeric single strand overhangs and t-loop junctions in vitro. J. Biol. Chem. 2002, 277, 11625–11628. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Muselikova-Polanska, E.; Pospisilova, S.; Strauss, F. High-affinity binding of tumor-suppressor protein p53 and HMGB1 to hemicatenated DNA loops. Biochemistry 2004, 43, 7215–7225. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Coufal, J.; Jagelska, E.B.; Liao, J.C.C.; Brazda, V. Preferential binding of p53 tumor suppressor to p21 promoter sites that contain inverted repeats capable of forming cruciform structure. Biochem. Biophys. Res. Commun. 2013, 441, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Čechová, J.; Coufal, J.; Jagelská, E.B.; Fojta, M.; Brázda, V. p73, like its p53 homolog, shows preference for inverted repeats forming cruciforms. PLoS ONE 2018, 13, e0195835. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Cechova, J.; Battistin, M.; Coufal, J.; Jagelska, E.B.; Raimondi, I.; Inga, A. The structure formed by inverted repeats in p53 response elements determines the transactivation activity of p53 protein. Biochem. Biophys. Res. Commun. 2017, 483, 516–521. [Google Scholar] [CrossRef]
- Petr, M.; Helma, R.; Polaskova, A.; Krejci, A.; Dvorakova, Z.; Kejnovska, I.; Navratilova, L.; Adamik, M.; Vorlickova, M.; Brazdova, M. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016, 36, e00397. [Google Scholar] [CrossRef]
- Adamik, M.; Kejnovska, I.; Bazantova, P.; Petr, M.; Renciuk, D.; Vorlickova, M.; Brazdova, M. p53 binds human telomeric G-quadruplex in vitro. Biochimie 2016, 128, 83–91. [Google Scholar] [CrossRef]
- Lee, S.-H.; Siaw, G.E.-L.; Willcox, S.; Griffith, J.D.; Hsieh, T.-S. Synthesis and dissolution of hemicatenanes by type IA DNA topoisomerases. Proc. Natl. Acad. Sci. USA 2013, 110, E3587–E3594. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Lee, S.-J.; No, Y.R.; Dang, D.T.; Dang, L.H.; Yang, V.W.; Shim, H.; Yun, C.C. Regulation of Hypoxia-inducible Factor 1 alpha (HIF-1 alpha) by Lysophosphatidic Acid Is Dependent on Interplay between p53 and Kruppel-like Factor 5. J. Biol. Chem. 2013, 288, 25244–25253. [Google Scholar] [CrossRef] [PubMed]
- Stros, M.; Ozaki, T.; Bacikova, A.; Kageyama, H.; Nakagawara, A. HMGB1 and HMGB2 cell-specifically down-regulate the p53- and p73- dependent sequence-specific transactivation from the human Bax gene promoter. J. Biol. Chem. 2002, 277, 7157–7164. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Kar, A.; Willcox, S.; Griffith, J.D. Transcription of telomeric DNA leads to high levels of homologous recombination and t-loops. Nucleic Acids Res. 2016, 44, 9369–9380. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef] [PubMed]
- Tutton, S.; Azzam, G.A.; Stong, N.; Vladimirova, O.; Wiedmer, A.; Monteith, J.A.; Beishline, K.; Wang, Z.; Deng, Z.; Riethman, H.; et al. Subtelomeric p53 binding prevents accumulation of DNA damage at human telomeres. EMBO J. 2016, 35, 193–207. [Google Scholar] [CrossRef]
- Lieberman, P.M. Retrotransposon-derived p53 binding sites enhance telomere maintenance and genome protection. Bioessays 2016, 38, 943–949. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem 1995, 64, 65–95. [Google Scholar] [CrossRef]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef]
- Thomas, T.J.; Faaland, C.A.; Gallo, M.A.; Thomas, T. Suppression of c-myc oncogene expression by a polyamine-complexed triplex forming oligonucleotide in MCF-7 breast cancer cells. Nucleic Acids Res. 1995, 23, 3594–3599. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Y.; Zhang, W. G-quadruplex structures and their interaction diversity with ligands. ChemMedChem 2014, 9, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Day, H.A.; Pavlou, P.; Waller, Z.A.E. i-Motif DNA: Structure, stability and targeting with ligands. Bioorg. Med. Chem. 2014, 22, 4407–4418. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Maiti, S. A thermodynamic overview of naturally occurring intramolecular DNA quadruplexes. Nucleic Acids Res. 2008, 36, 5610–5622. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G.N. Quadruplex DNA crystal structures and drug design. Biochimie 2008, 90, 1184–1196. [Google Scholar] [CrossRef]
- Neidle, S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- Haronikova, L.; Coufal, J.; Kejnovska, I.; Jagelska, E.B.; Fojta, M.; Dvorakova, P.; Muller, P.; Vojtesek, B.; Brazda, V. IFI16 Preferentially Binds to DNA with Quadruplex Structure and Enhances DNA Quadruplex Formation. PLoS ONE 2016, 11, e0157156. [Google Scholar] [CrossRef]
- Kharel, P.; Balaratnam, S.; Beals, N.; Basu, S. The role of RNA G-quadruplexes in human diseases and therapeutic strategies. Wiley Interdiscip. Rev. RNA 2019, e1568. [Google Scholar] [CrossRef]
- Gomes, A.S.; Trovão, F.; Andrade Pinheiro, B.; Freire, F.; Gomes, S.; Oliveira, C.; Domingues, L.; Romão, M.J.; Saraiva, L.; Carvalho, A.L. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. Int. J. Mol. Sci. 2018, 19, 1184. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.; Pearsall, I.; Yeudall, A.; Deb, S.P.; Deb, S. p53: Its mutations and their impact on transcription. Subcell. Biochem. 2014, 85, 71–90. [Google Scholar] [PubMed]
- Brazda, V.; Muller, P.; Brozkova, K.; Vojtesek, B. Restoring wild-type conformation and DNA-binding activity of mutant p53 is insufficient for restoration of transcriptional activity. Biochem. Biophys. Res. Commun. 2006, 351, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Viadiu, H.; Fronza, G.; Inga, A. Structural studies on mechanisms to activate mutant p53. Subcell. Biochem. 2014, 85, 119–132. [Google Scholar]
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef]
- Brazdova, M.; Quante, T.; Toegel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.X.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.J.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun. 2000, 279, 383–390. [Google Scholar] [CrossRef]
- Brazda, V.; Haronikova, L.; Liao, J.C.C.; Fojta, M. DNA and RNA Quadruplex-Binding Proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef]
- Ramos, E.M.; Gillis, T.; Mysore, J.S.; Lee, J.M.; Alonso, I.; Gusella, J.F.; Smoller, J.W.; Sklar, P.; MacDonald, M.E.; Perlis, R.H. Prevalence of Huntington’s disease gene CAG trinucleotide repeat alleles in patients with bipolar disorder. Bipolar Disord. 2015, 17, 403–408. [Google Scholar] [CrossRef]
- Den Dunnen, W.F.A. Trinucleotide repeat disorders. In Handbook of Clinical Neurology; Elsevier B.V.: Oxford, UK, 2017; Volume 145, pp. 383–391. [Google Scholar]
- Pearson, C.E.; Sinden, R.R. Trinucleotide repeat DNA structures: Dynamic mutations from dynamic DNA. Curr. Opin. Struct. Biol. 1998, 8, 321–330. [Google Scholar] [CrossRef]
- Beaver, J.M.; Lai, Y.; Rolle, S.J.; Liu, Y. Proliferating cell nuclear antigen prevents trinucleotide repeat expansions by promoting repeat deletion and hairpin removal. DNA Repair 2016, 48, 17–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pearson, C.E.; Zorbas, H.; Price, G.B.; ZannisHadjopoulos, M. Inverted repeats, stem-loops, and cruciforms: Significance for initiation of DNA replication. J. Cell. Biochem. 1996, 63, 1–22. [Google Scholar] [CrossRef]
- Brázda, V.; Coufal, J.; Liao, J.C.C.; Arrowsmith, C.H. Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem. Biophys. Res. Commun. 2012, 422, 716–720. [Google Scholar] [CrossRef] [PubMed]
- van Holde, K.; Zlatanova, J. Unusual DNA structures, chromatin and transcription. Bioessays 1994, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Rohaly, G.; Heinrichs, S.; Gimnopoulos, D.; Meissner, H.; Deppert, W. Influence of promoter DNA topology on sequence-specific DNA binding and transactivation by tumor suppressor p53. Oncogene 1999, 18, 7310–7318. [Google Scholar] [CrossRef]
- Saramaki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D-3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef]
- Jett, S.D.; Cherny, D.I.; Subramaniam, V.; Jovin, T.M. Scanning force microscopy of the complexes of p53 core domain with supercoiled DNA. J. Mol. Biol. 2000, 299, 585–592. [Google Scholar] [CrossRef]
- Cherny, D.I.; Striker, G.; Subramaniam, V.; Jett, S.D.; Palecek, E.; Jovin, T.M. DNA bending due to specific p53 and p53 core domain-DNA interactions visualized by electron microscopy. J. Mol. Biol. 1999, 294, 1015–1026. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.-J.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; LoVerso, P.R.; Fisk, J.N.; Zhurkin, V.B.; Cui, F. p53 binding sites in normal and cancer cells are characterized by distinct chromatin context. Cell Cycle 2017, 16, 2073–2085. [Google Scholar] [CrossRef] [PubMed]
- Nabilsi, N.H.; Ryder, D.J.; Peraza-Penton, A.C.; Poudyal, R.; Loose, D.S.; Kladde, M.P. Local depletion of DNA methylation identifies a repressive p53 regulatory region in the NEK2 promoter. J. Biol. Chem. 2013, 288, 35940–35951. [Google Scholar] [CrossRef] [PubMed]
- Sammons, M.A.; Zhu, J.; Drake, A.M.; Berger, S.L. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015, 25, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Buck, M.J. Defining TP53 pioneering capabilities with competitive nucleosome binding assays. Genome Res. 2019, 29, 107–115. [Google Scholar] [CrossRef]
- Taube, J.H.; Allton, K.; Duncan, S.A.; Shen, L.; Barton, M.C. Foxa1 functions as a pioneer transcription factor at transposable elements to activate Afp during differentiation of embryonic stem cells. J. Biol. Chem. 2010, 285, 16135–16144. [Google Scholar] [CrossRef]
- Zhabinskaya, D.; Benham, C.J. Competitive superhelical transitions involving cruciform extrusion. Nucleic Acids Res. 2013, 41, 9610–9621. [Google Scholar] [CrossRef]
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: The interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730. [Google Scholar] [CrossRef]
- Krasilnikov, A.S.; Podtelezhnikov, A.; Vologodskii, A.; Mirkin, S.M. Large-scale effects of transcriptional DNA supercoiling in vivo. J. Mol. Biol. 1999, 292, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Guilbaud, G.; Murat, P.; Papadopoulou, C.; Sarkies, P.; Prioleau, M.-N.; Balasubramanian, S.; Sale, J.E. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014, 33, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Murat, P.; Recolin, B.; Campbell, B.C.; Maiter, A.; Sale, J.E.; Balasubramanian, S. Local epigenetic reprogramming induced by G-quadruplex ligands. Nat. Chem. 2017, 9, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Leach, D.R. DNA palindromes adopt a methylation-resistant conformation that is consistent with DNA cruciform or hairpin formation in vivo. J. Mol. Biol. 1995, 252, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Jara-Espejo, M.; Peres Line, S.R. DNA G-quadruplex stability, position and chromatin accessibility are associated with CpG island methylation. FEBS J. 2019. [Google Scholar] [CrossRef]
- Schlereth, K.; Heyl, C.; Krampitz, A.-M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the p53 cistrome—DNA binding cooperativity dissects p53′s tumor suppressor functions. PLoS Genet. 2013, 9, e1003726. [Google Scholar] [CrossRef]
- Stiewe, T.; Haran, T.E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updat. 2018, 38, 27–43. [Google Scholar] [CrossRef]
- Ohtsuka, J.; Oshima, H.; Ezawa, I.; Abe, R.; Oshima, M.; Ohki, R. Functional loss of p53 cooperates with the in vivo microenvironment to promote malignant progression of gastric cancers. Sci. Rep. 2018, 8, 2291. [Google Scholar] [CrossRef]
- Nakayama, M.; Sakai, E.; Echizen, K.; Yamada, Y.; Oshima, H.; Han, T.-S.; Ohki, R.; Fujii, S.; Ochiai, A.; Robine, S.; et al. Intestinal cancer progression by mutant p53 through the acquisition of invasiveness associated with complex glandular formation. Oncogene 2017, 36, 5885–5896. [Google Scholar] [CrossRef]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brázda, V.; Fojta, M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int. J. Mol. Sci. 2019, 20, 5605. https://doi.org/10.3390/ijms20225605
Brázda V, Fojta M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. International Journal of Molecular Sciences. 2019; 20(22):5605. https://doi.org/10.3390/ijms20225605
Chicago/Turabian StyleBrázda, Václav, and Miroslav Fojta. 2019. "The Rich World of p53 DNA Binding Targets: The Role of DNA Structure" International Journal of Molecular Sciences 20, no. 22: 5605. https://doi.org/10.3390/ijms20225605
APA StyleBrázda, V., & Fojta, M. (2019). The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. International Journal of Molecular Sciences, 20(22), 5605. https://doi.org/10.3390/ijms20225605