A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
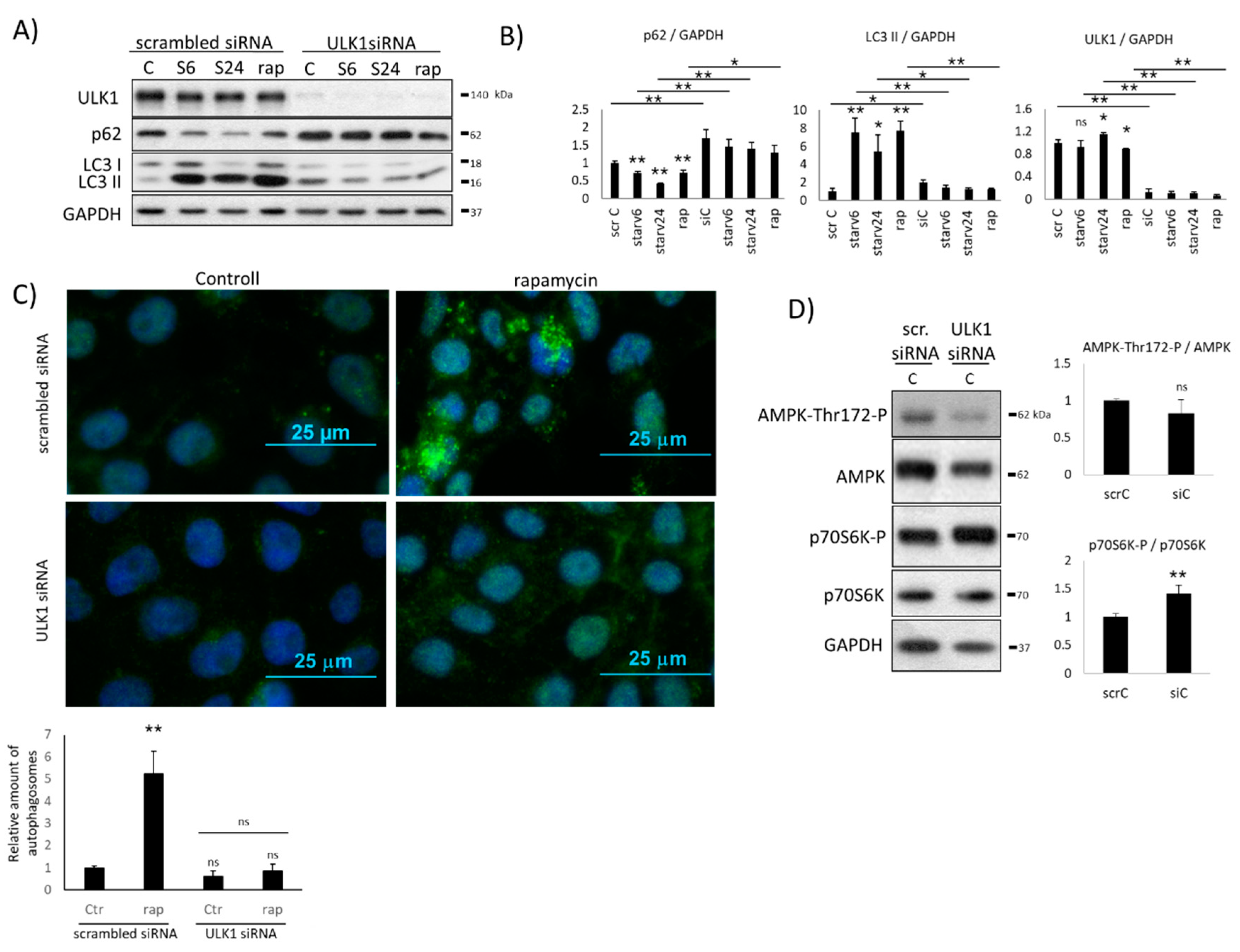
2.1. Downregulation of ULK1 Blocks AMPK Induction
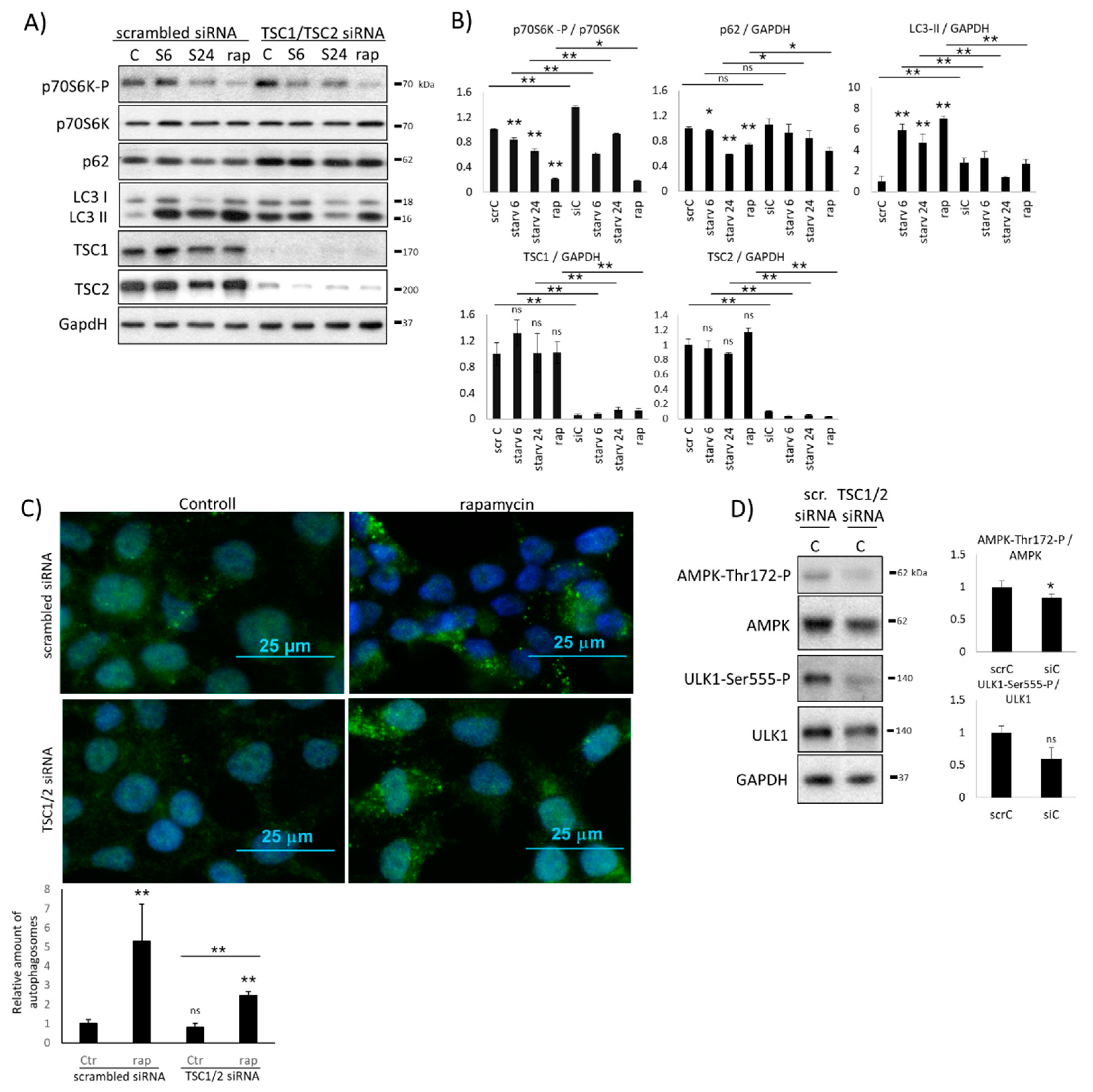
2.2. Hyper-Activation of mTOR Blocks AMPK Induction
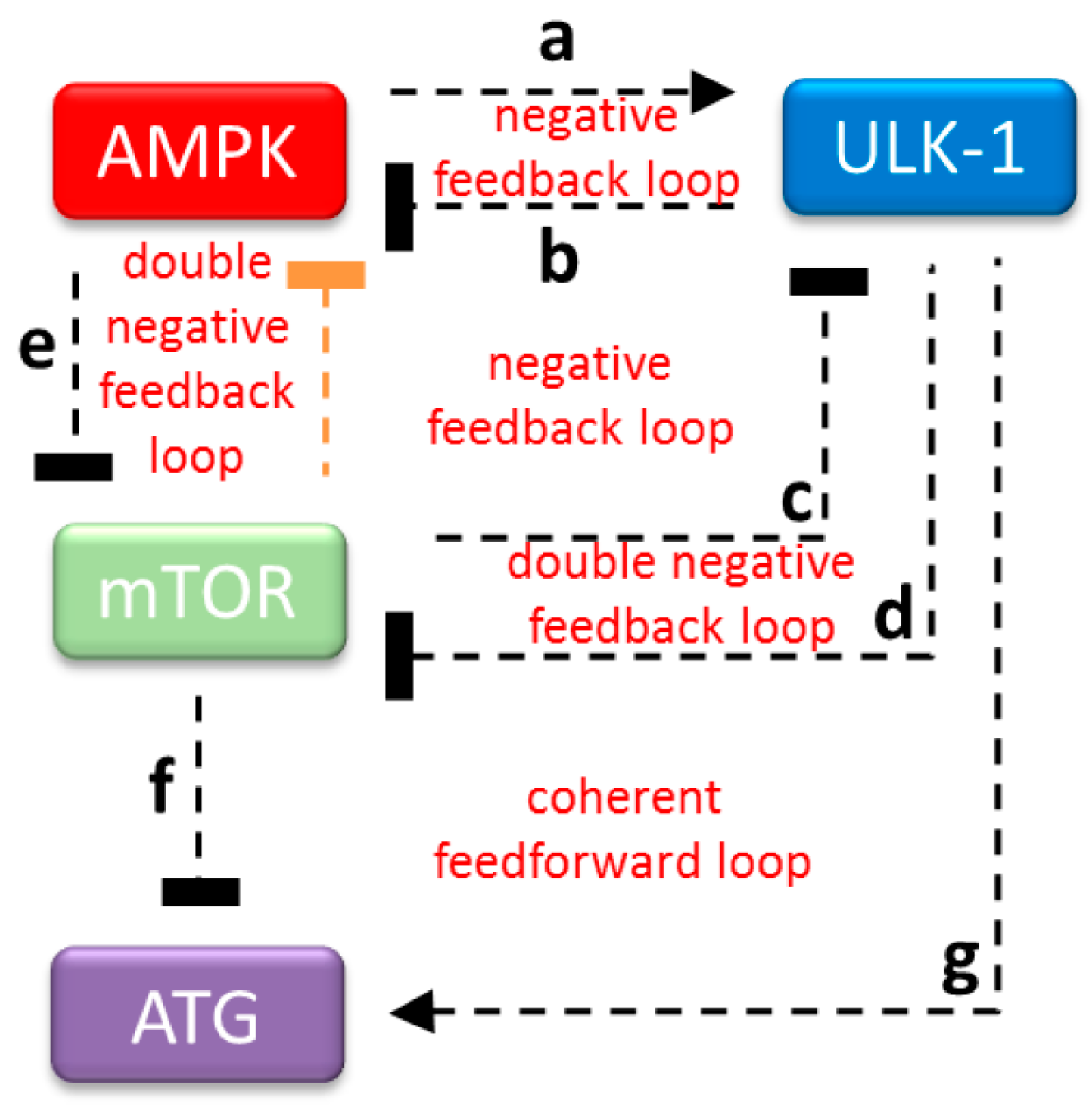
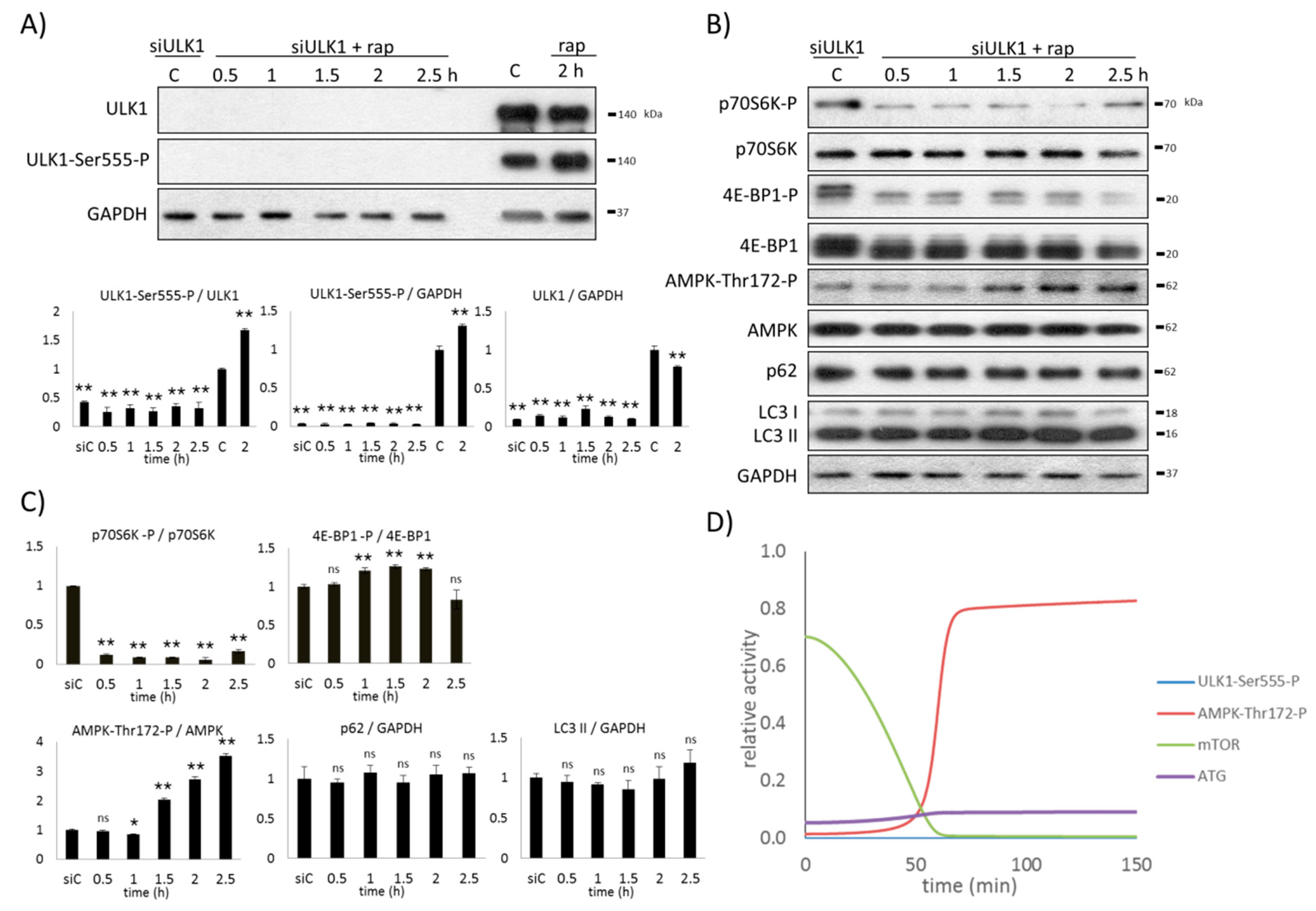
2.3. The Proper Dynamical Characteristic of ULK1 Depletion Could Be Managed by a Double Negative Feedback Loop between mTOR and AMPK
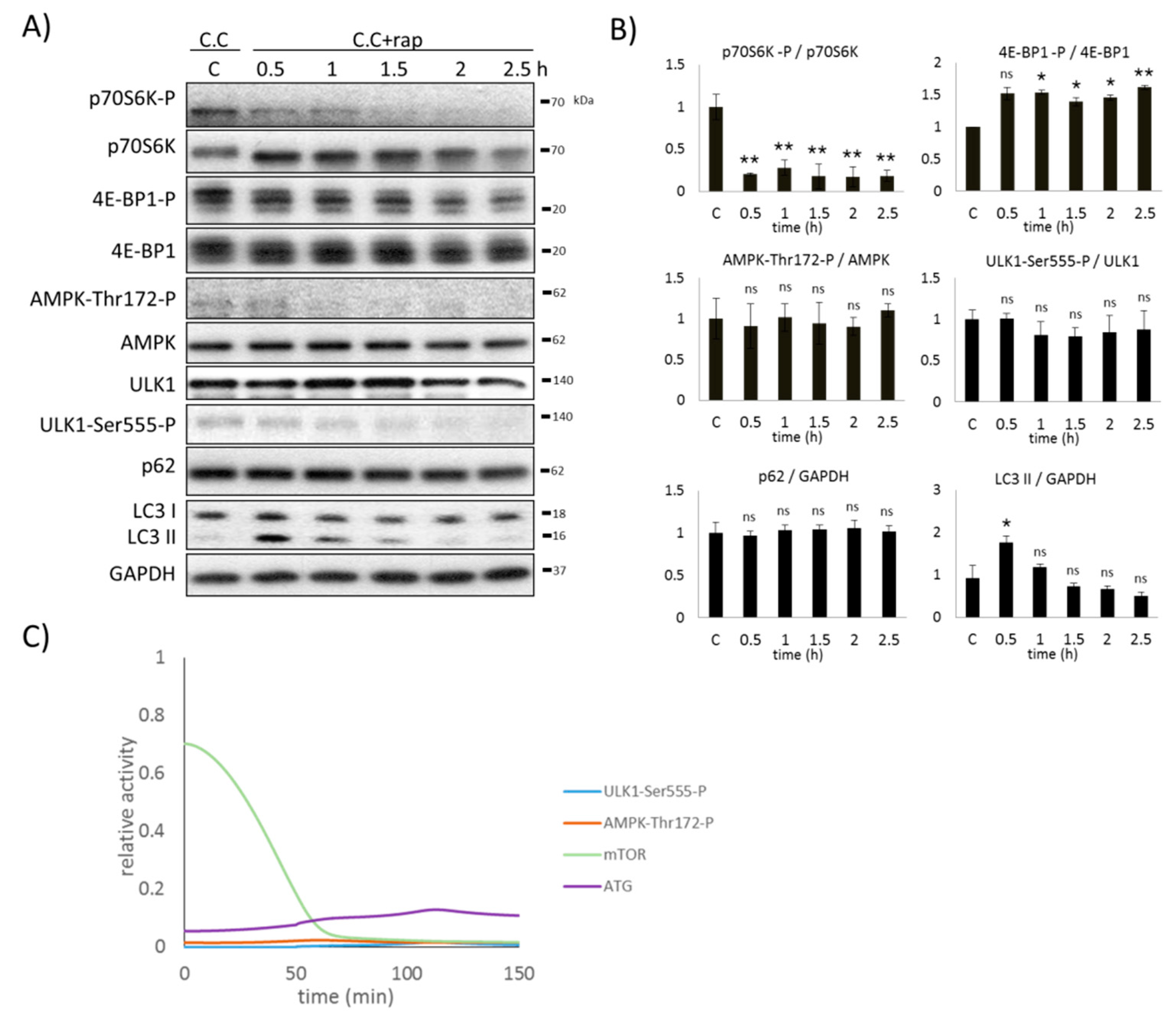
2.4. AMPK Activation Always Precedes ULK1 Induction during Autophagic Stress Response
2.5. AMPK Is Not Sufficient to Induce Autophagy during mTOR-Downregulation
2.6. AMPK Is Essential to Induce Autophagy during mTOR-Downregulation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Maintenance
4.3. RNA Interference
4.4. SDS-PAGE and Western Blot Analysis
4.5. Immunofluorescence
4.6. Mathematical Modeling
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mTOR | Mammalian target of rapamycin |
| AMPK | 5′ AMP-activated protein kinase |
| rap | Rapamycin |
| CC | Compound C |
| s6 | 6 h starvation |
| s24 | 24 h starvation |
References
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Carling, D.; Gamblin, S.J. AMP-activated protein kinase: Also regulated by ADP? Trends Biochem. Sci. 2011, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Tamargo-Gomez, I.; Marino, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef]
- Watanabe, R.; Wei, L.; Huang, J. mTOR Signaling, Function, Novel Inhibitors, and Therapeutic Targets. J. Nucl. Med. 2011, 52, 497–500. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Jaattela, M. AMP-activated protein kinase: A universal regulator of autophagy? Autophagy 2007, 3, 381–383. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- Kung, C.P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M. Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [PubMed]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Loffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, P.; Martin, K.R.; MacKeigan, J.P.; Hlavacek, W.S.; Lipniacki, T. Computational analysis of an autophagy/translation switch based on mutual inhibition of MTORC1 and ULK1. PLoS ONE 2015, 10, e0116550. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.H. Identification of an AMPK phosphorylation site in Drosophila TSC2 (gigas) that regulate cell growth. Int. J. Mol. Sci. 2015, 16, 7015–7026. [Google Scholar] [CrossRef]
- Li, H.; Lee, J.; He, C.; Zou, M.H.; Xie, Z. Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E197–E209. [Google Scholar] [CrossRef]
- Green, A.S.; Chapuis, N.; Maciel, T.T.; Willems, L.; Lambert, M.; Arnoult, C.; Boyer, O.; Bardet, V.; Park, S.; Foretz, M.; et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood 2010, 116, 4262–4273. [Google Scholar] [CrossRef]
- Oosterman, J.E.; Belsham, D.D. Glucose Alters Per2 Rhythmicity Independent of AMPK, Whereas AMPK Inhibitor Compound C Causes Profound Repression of Clock Genes and AgRP in mHypoE-37 Hypothalamic Neurons. PLoS ONE 2016, 11, e0146969. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Kosztelnik, M.; Kurucz, A.; Papp, D.; Jones, E.; Sigmond, T.; Barna, J.; Traka, M.H.; Lorincz, T.; Szarka, A.; Banhegyi, G.; et al. Suppression of AMPK/aak-2 by NRF2/SKN-1 down-regulates autophagy during prolonged oxidative stress. FASEB J. 2018, 33, 2372–2387. [Google Scholar] [CrossRef]
- Kapuy, O.; Papp, D.; Vellai, T.; Bánhegyi, G.; Korcsmáros, T. Systems-Level Feedbacks of NRF2 Controlling Autophagy upon Oxidative Stress Response. Antioxidants (Basel) 2018, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharm. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharm. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Kubisch, J.; Türei, D.; Földvári-Nagy, L.; Dunai, Z.A.; Zsákai, L.; Varga, M.; Vellai, T.; Csermely, P.; Korcsmáros, T. Complex regulation of autophagy in cancer-integrated approaches to discover the networks that hold a double-edged sword. Semin. Cancer Biol. 2013, 23, 252–261. [Google Scholar] [CrossRef]
- Kaplan, D.; Glass, L. Understanding Nonlinear Dynamics; Springer: New York, NY, USA, 1995. [Google Scholar]
- Strogatz, S.H. Nonlinear Dynamics and Chaos; Addison-Wesley Co.: Reading, MA, USA, 1994. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. Int. J. Mol. Sci. 2019, 20, 5543. https://doi.org/10.3390/ijms20225543
Holczer M, Hajdú B, Lőrincz T, Szarka A, Bánhegyi G, Kapuy O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. International Journal of Molecular Sciences. 2019; 20(22):5543. https://doi.org/10.3390/ijms20225543
Chicago/Turabian StyleHolczer, Marianna, Bence Hajdú, Tamás Lőrincz, András Szarka, Gábor Bánhegyi, and Orsolya Kapuy. 2019. "A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress" International Journal of Molecular Sciences 20, no. 22: 5543. https://doi.org/10.3390/ijms20225543
APA StyleHolczer, M., Hajdú, B., Lőrincz, T., Szarka, A., Bánhegyi, G., & Kapuy, O. (2019). A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. International Journal of Molecular Sciences, 20(22), 5543. https://doi.org/10.3390/ijms20225543