Impact of RNA Virus Evolution on Quasispecies Formation and Virulence


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Picornaviridae
2.1. Poliovirus (PV)
2.2. Enterovirus 71 (EV-A71)
3. Flaviviridae
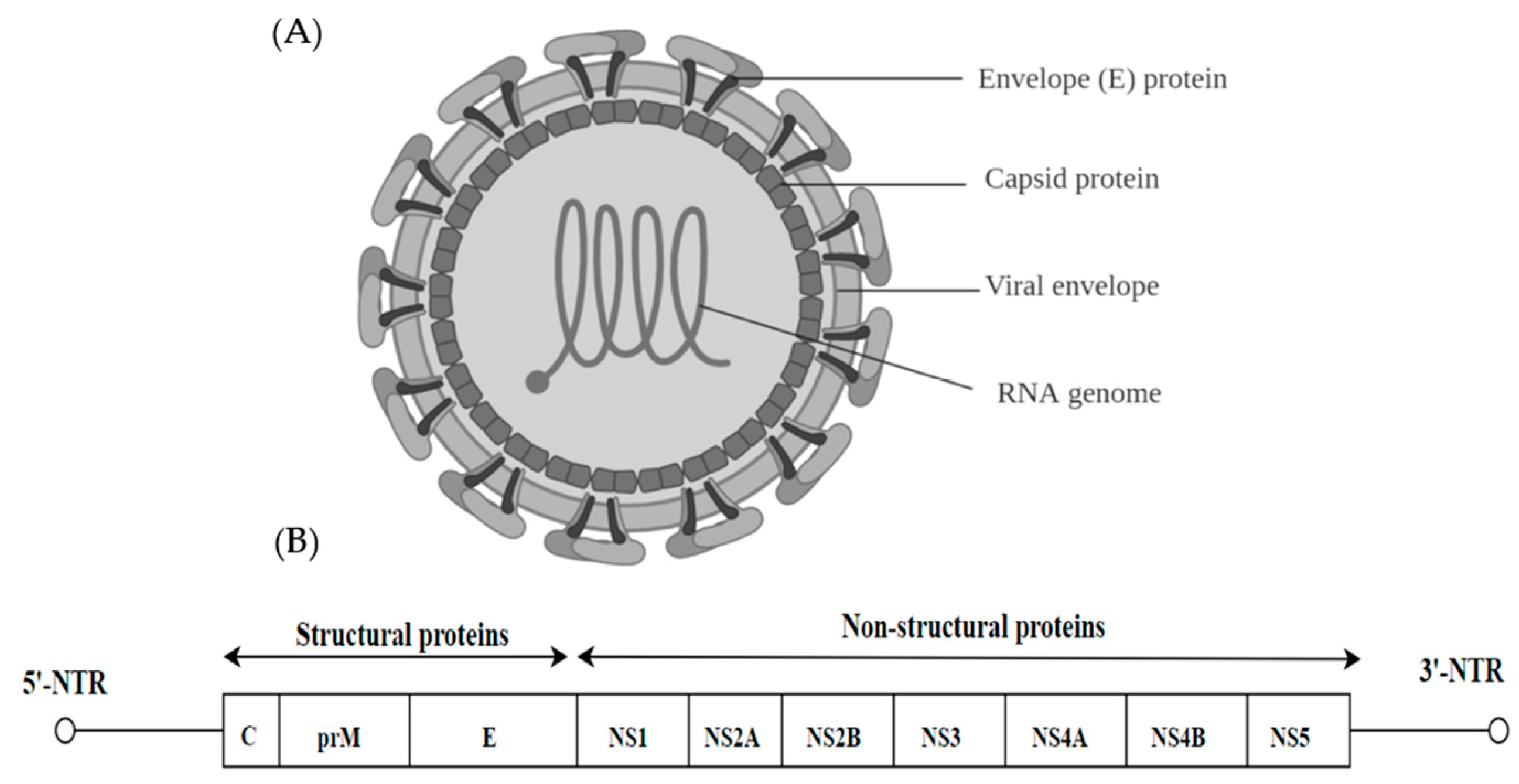
3.1. Dengue Virus (DENV)
3.2. Zika Virus (ZIKV)
3.3. West Nile Virus (WNV)
4. Togaviridae
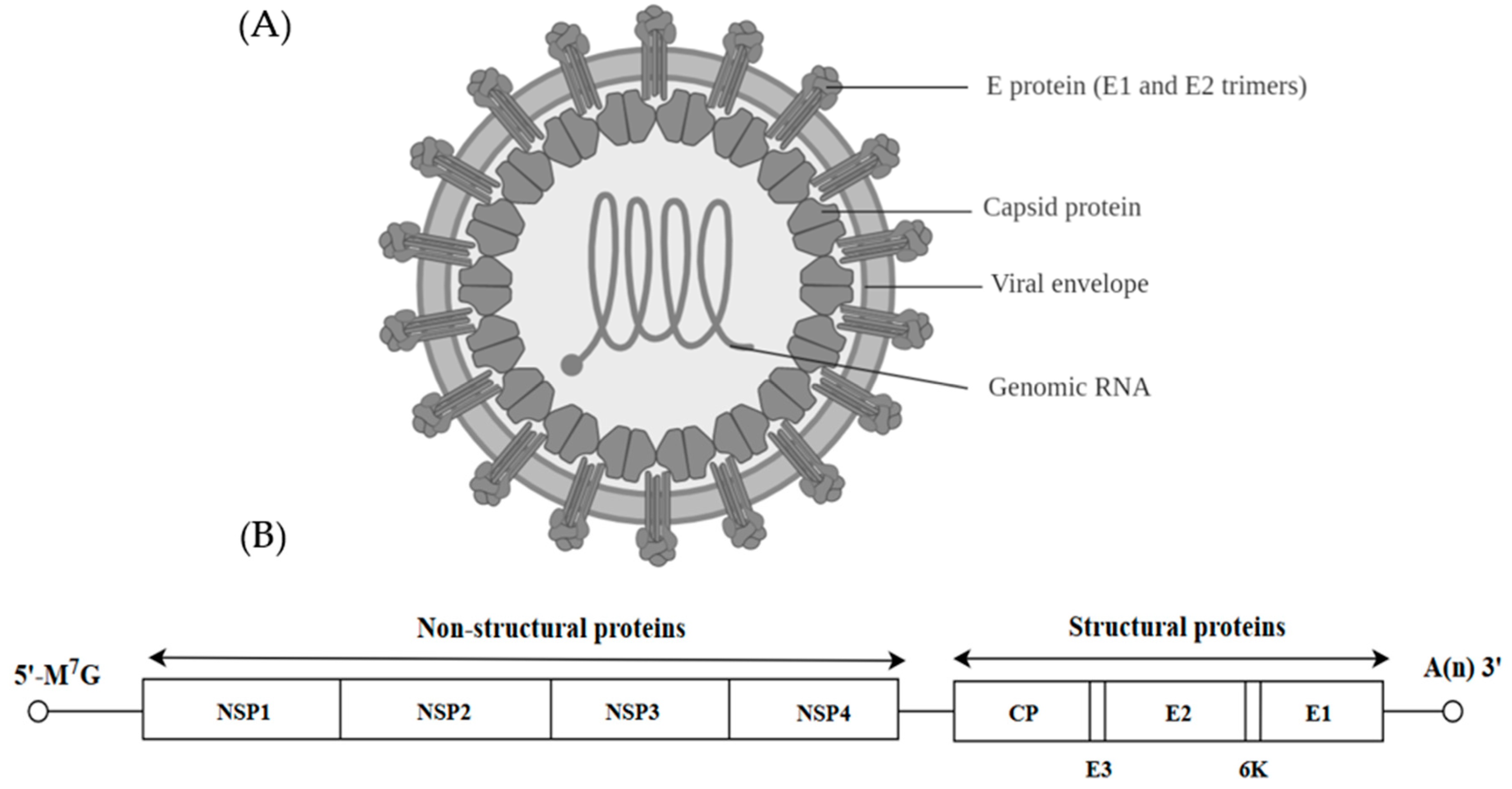
Chikungunya Virus (CHIKV)
5. Filoviridae
Ebola Virus (EboV)
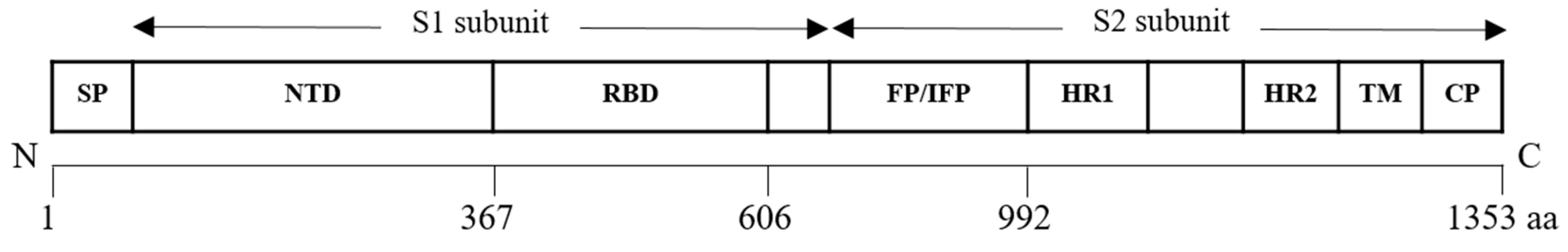
6. Coronaviridae
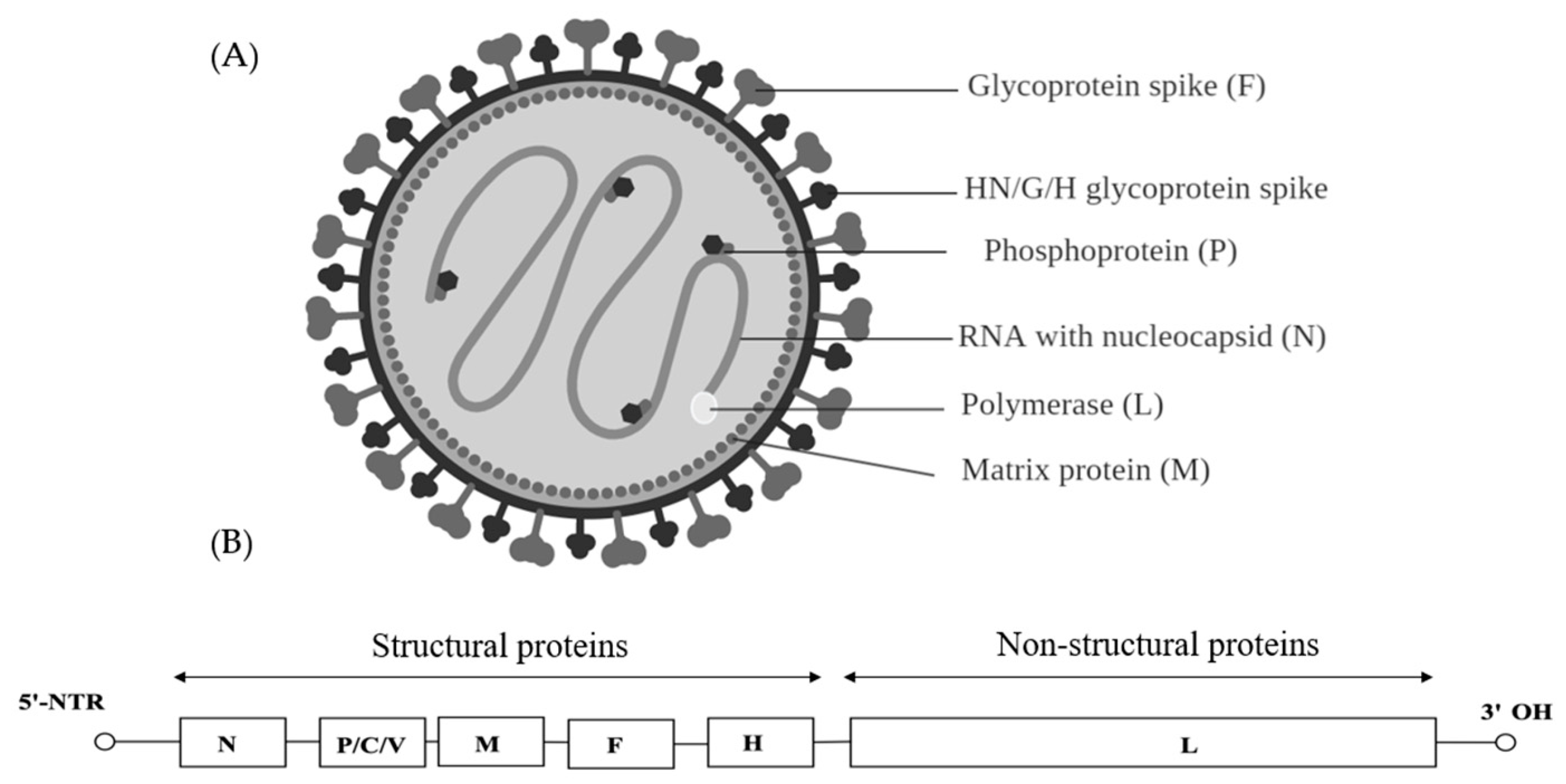
7. Paramyxoviridae
7.1. Measles Virus (MeV)
7.2. Mumps
7.3. Newcastle Disease Virus (NDV)
8. Pneumoviridae
Respiratory Syncytial Virus (RSV)
9. Orthomyxoviridae
Influenza Virus (IV)
10. Hepadnaviridae
Hepatitis B Virus (HBV)
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Institute of Medicine, Committee on Emerging Microbial Threats to Health. Emerging Infections: Microbial Threats to Health in the United States; Oaks, S.C., Jr., Shope, R.E., Lederberg, J., Eds.; National Academies Press: Washington, DC, USA, 1992. [Google Scholar]
- Gerrish, P.J.; García-Lerma, J.G. Mutation rate and the efficacy of antimicrobial drug treatment. Lancet Infect. Dis. 2003, 3, 28–32. [Google Scholar] [CrossRef]
- Domingo, E. RNA virus evolution and the control of viral disease. Prog. Drug. Res. 1989, 33, 93–133. [Google Scholar] [PubMed]
- Domingo, E.; Baranowski, E.; Ruiz-Jarabo, C.M.; Martín-Hernández, A.M.; Sáiz, J.C.; Escarmís, C. Quasispecies structure and persistence of RNA viruses. Emerg. Infect. Dis. 1998, 4, 521. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Domingo, E.; Sabo, D.; Taniguchi, T.; Weissmann, C. Nucleotide sequence heterogeneity of an RNA phage population. Cell 1978, 13, 735–744. [Google Scholar] [CrossRef]
- Batschelet, E.; Domingo, E.; Weissmann, C. The proportion of revertant and mutant phage growing in a population, as a function of mutation and growth rate. Gene 1976, 1, 27–32. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. The hypercycle. Naturwissenschaften 1978, 65, 7–41. [Google Scholar] [CrossRef]
- Drake, J.W. Spontaneous mutation: Comparative rates of spontaneous mutation. Nature 1969, 221, 1132. [Google Scholar] [CrossRef]
- Steinhauer, D.A.; Holland, J. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 1987, 41, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Schuster, P. Quasispecies: From Theory to Experimental Systems; Springer: Basel, Switzerland, 2016; p. 392. [Google Scholar]
- Pfeiffer, J.K.; Kirkegaard, K. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc. Natl. Acad. Sci. USA 2006, 103, 5520–5525. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Hooper, D.C.; Carbaugh, H.; Fu, Z.F.; Koprowski, H.; Dietzschold, B. Rabies virus quasispecies: Implications for pathogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 3152–3156. [Google Scholar] [CrossRef] [PubMed]
- Donohue, R.C.; Pfaller, C.K.; Cattaneo, R. Cyclical adaptation of measles virus quasispecies to epithelial and lymphocytic cells: To V or not to V. PLoS Pathog. 2019, 15, e1007605. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.; Hovi, T.; Hyypiä, T.; King, A.; Lindberg, A.M.; Pallansch, M.; Palmenberg, A.; Simmonds, P.; Skern, T.; Stanway, G. Family-Picornaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Brown, B.A.; Pallansch, M.A. Complete nucleotide sequence of Enterovirus 71 is distinct from Poliovirus. Virus Res. 1995, 39, 195–205. [Google Scholar] [CrossRef]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis and control of Enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- World Health Organization. Poliomyelitis. Available online: https://www.who.int/news-room/fact-sheets/detail/poliomyelitis (accessed on 23 July 2019).
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]
- Moratorio, G.; Henningsson, R.; Barbezange, C.; Carrau, L.; Bordería, A.V.; Blanc, H.; Beaucourt, S.; Poirier, E.Z.; Vallet, T.; Boussier, J. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol. 2017, 2, 17088. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.J.; Lennette, E.H.; Ho, H.H. An apparently new enterovirus isolated from patients with disease of the central nervous system. J. Infect. Dis. 1974, 129, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, Y.; Nakano, S.; Yamaoka, K.; Takami, S. Outbreaks of hand, foot and mouth disease by enterovirus 71. High incidence of complication disorders of central nervous system. Arch. Dis. Child. 1980, 55, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.P., Jr.; Baden, L.; Pallansch, M.A.; Anderson, L.J. Enterovirus 71 infections and neurologic disease—United States, 1977–1991. J. Infect. Dis. 1994, 169, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y. Hand, foot and mouth disease in China, 2008–12: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar] [CrossRef]
- Mandary, M.; Poh, C. Changes in the EV-A71 genome through recombination and spontaneous mutations: impact on virulence. Viruses 2018, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-W.; Huang, Y.-H.; Tsai, H.-P.; Kuo, P.-H.; Wang, S.-M.; Liu, C.-C.; Wang, J.-R. A selective bottleneck shapes the evolutionary mutant spectra of Enterovirus A71 during viral dissemination in humans. J. Virol. 2017, 91, e01062-17. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Collett, M.; Gould, E.; Heinz, F.; Meyers, G.; Monath, T.; Pletnev, A.; Rice, C.; Stiasny, K. Family- Flaviviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 1003–1020. [Google Scholar]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Gurugama, P.; Garg, P.; Perera, J.; Wijewickrama, A.; Seneviratne, S.L. Dengue viral infections. Indian J. Dermatol. 2010, 55, 68. [Google Scholar]
- World Health Organization. Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 15 April 2019).
- Henchal, E.A.; Putnak, J.R. The Dengue viruses. Clin. Microbiol. Rev. 1990, 3, 376–396. [Google Scholar] [CrossRef]
- Whitehorn, J.; Simmons, C.P. The pathogenesis of Dengue. Vaccine 2011, 29, 7221–7228. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.C.M.; Tang, C.K.; Norton, D.C.; Gan, E.S.; Tan, H.C.; Sun, B.; Syenina, A.; Yousuf, A.; Ong, X.M.; Kamaraj, U.S.; et al. Molecular determinants of plaque size as an indicator of Dengue virus attenuation. Sci. Rep. 2016, 6, 26100. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-K.; Lin, S.-R.; Lee, C.-M.; King, C.-C.; Chang, S.-C. Dengue type 3 virus in plasma is a population of closely related genomes: quasispecies. J. Virol. 2002, 76, 4662–4665. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, P.; Wang, C.; Trivedi, S.B.; Eswarappa, M.; Montoya, M.; Balmaseda, A.; Harris, E. Intrahost selection pressures drive rapid Dengue virus microevolution in acute human infections. Cell Host Microbe 2017, 22, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Love, H. Investigation of Denugue Virus Envelope Gene Quasispecies Variation in Patient Samples: Implications for Virus Virulence and Disease Pathognesis. Ph.D. Thesis, Cranfield University, Cranfield, UK, 2011. [Google Scholar]
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika Virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Smithburn, K.C.; Taylor, R.M.; Rizk, F.; Kader, A. Immunity to certain arthropod-borne viruses among indigenous residents of Egypt. Am. J. Trop. Med. Hyg. 1954, 3, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Smithburn, K.C. Neutralizing antibodies against certain recently isolated viruses in the sera of human beings residing in East Africa. J. Immunol. 1952, 69, 223–234. [Google Scholar] [PubMed]
- Smithburn, K.C.; Kerr, J.A.; Gatne, P.B. Neutralizing antibodies against certain viruses in the sera of residents of India. J. Immunol. 1954, 72, 248–257. [Google Scholar] [PubMed]
- Pond, W.L. Arthropod-borne virus antibodies in sera from residents of South-East Asia. Trans. R. Soc. Trop. Med. Hyg. 1963, 57, 364–371. [Google Scholar] [CrossRef]
- Smithburn, K.C. Neutralizing antibodies against arthropod-borne viruses in the sera of long-time residents of Malaya and Borneo. Am. J. Hyg. 1954, 59, 157–163. [Google Scholar]
- Kato, F.; Tajima, S.; Nakayama, E.; Kawai, Y.; Taniguchi, S.; Shibasaki, K.; Taira, M.; Maeki, T.; Lim, C.K.; Takasaki, T. Characterization of large and small-plaque variants in the Zika virus clinical isolate ZIKV/Hu/S36/Chiba/2016. Sci. Rep. 2017, 7, 16160. [Google Scholar] [CrossRef] [PubMed]
- van Boheemen, S.; Tas, A.; Anvar, S.Y.; van Grootveld, R.; Albulescu, I.C.; Bauer, M.P.; Feltkamp, M.C.; Bredenbeek, P.J.; van Hemert, M.J. Quasispecies composition and evolution of a typical Zika virus clinical isolate from Suriname. Sci. Rep. 2017, 7, 2368. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.L.; Marfin, A.A.; Lanciotti, R.S.; Gubler, D.J. West Nile Virus. Lancet Infect. Dis. 2002, 2, 519–529. [Google Scholar] [CrossRef]
- Turell, M.J.; Sardelis, M.R.; Dohm, D.J.; O’Guinn, M.L. Potential North American vectors of West Nile virus. Ann. N. Y. Acad. Sci. 2001, 951, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Moudy, R.M.; Dupuis, A.P.; Ngo, K.A.; Maffei, J.G.; Jerzak, G.V.; Franke, M.A.; Kauffman, E.B.; Kramer, L.D. Characterization of a small plaque variant of West Nile virus isolated in New York in 2000. Virology 2007, 367, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Dridi, M.; Rosseel, T.; Orton, R.; Johnson, P.; Lecollinet, S.; Muylkens, B.; Lambrecht, B.; Van Borm, S. Next-generation sequencing shows West Nile virus quasispecies diversification after a single passage in a carrion crow (Corvus corone) in vivo infection model. J. Gen. Virol. 2015, 96, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.; Huang, H.; Roehrig, J.; Strauss, E.; Weaver, S. Family- Togaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 1103–1110. [Google Scholar]
- Wachtman, L.; Mansfield, K. Viral diseases of nonhuman primates. In Nonhuman Primates in Biomedical Research; Abee, C., Mansfield, K., Tardif, S., Morris, T., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 2, pp. 1–104. [Google Scholar]
- Weaver, S.C. Arrival of Chikungunya virus in the new world: prospects for spread and impact on public health. PLoS Negl. Trop. Dis. 2014, 8, e2921. [Google Scholar] [CrossRef] [PubMed]
- Cavrini, F.; Gaibani, P.; Pierro, A.; Rossini, G.; Landini, M.; Sambri, V. Chikungunya: an emerging and spreading arthropod-borne viral disease. J. Infect. Dev. Ctries. 2009, 3, 744–752. [Google Scholar] [CrossRef]
- Thiboutot, M.M.; Kannan, S.; Kawalekar, O.U.; Shedlock, D.J.; Khan, A.S.; Sarangan, G.; Srikanth, P.; Weiner, D.B.; Muthumani, K. Chikungunya: A potentially emerging epidemic? PLoS Negl. Trop. Dis. 2010, 4, e623. [Google Scholar] [CrossRef] [PubMed]
- Lemant, J.; Boisson, V.; Winer, A.; Thibault, L.; André, H.; Tixier, F.; Lemercier, M.; Antok, E.; Cresta, M.P.; Grivard, P. Serious acute chikungunya virus infection requiring intensive care during the Reunion Island outbreak in 2005–2006. Crit. Care Med. 2008, 36, 2536–2541. [Google Scholar] [CrossRef]
- Leparc-Goffart, I.; Nougairede, A.; Cassadou, S.; Prat, C.; de Lamballerie, X. Chikungunya in the Americas. Lancet 2014, 383, 514. [Google Scholar] [CrossRef]
- Josseran, L.; Paquet, C.; Zehgnoun, A.; Caillere, N.; Le Tertre, A.; Solet, J.-L.; Ledrans, M. Chikungunya disease outbreak, Reunion Island. Emerg. Infect. Dis. 2006, 12, 1994–1995. [Google Scholar] [CrossRef] [PubMed]
- Pulmanausahakul, R.; Roytrakul, S.; Auewarakul, P.; Smith, D.R. Chikungunya in Southeast Asia: Understanding the emergence and finding solutions. Int. J. Infect. Dis. 2011, 15, e671–e676. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, E.; Issac, A.; Nair, S.; Hariharan, R.; Janki, M.B.; Arathy, D.S.; Regu, R.; Mathew, T.; Anoop, M.; Niyas, K.P.; et al. Genetic characterization of 2006–2008 isolates of Chikungunya virus from Kerala, South India, by whole genome sequence analysis. Virus Genes 2009, 40, 14. [Google Scholar] [CrossRef] [PubMed]
- Sudeep, A.; Parashar, D. Chikungunya: an overview. J. Biosci. 2008, 33, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in Chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Shalauddin, A.; Lorraine, F.; Ricketts, R.P.; Trudy, C.; Karen, P.-E.; Babatunde, O. Chikungunya virus outbreak, Dominica, 2014. Emerging Infect. Dis. 2015, 21, 909. [Google Scholar]
- Sam, I.C.; Kümmerer, B.M.; Chan, Y.-F.; Roques, P.; Drosten, C.; AbuBakar, S. Updates on Chikungunya epidemiology, clinical disease and diagnostics. Vector Borne Zoonotic Dis. 2015, 15, 223–230. [Google Scholar] [CrossRef]
- Powers, A.M. Vaccine and therapeutic options to control Chikungunya virus. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Riemersma, K.K.; Steiner, C.; Singapuri, A.; Coffey, L.L. Chikungunya virus fidelity variants exhibit differential attenuation and population diversity in cell culture and adult mice. J. Virol. 2019, 93, e01606-18. [Google Scholar] [CrossRef]
- Lim, C.; Nishibori, T.; Watanabe, K.; Ito, M.; Kotaki, A.; Tanaka, K.; Kurane, I.; Takasaki, T. Chikungunya virus isolated from a returnee to Japan from Sri Lanka: isolation of two sub-strains with different characteristics. Am. J. Trop. Med. Hyg. 2009, 81, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Wasonga, C.; Inoue, S.; Rumberia, C.; Michuki, G.; Kimotho, J.; Ongus, J.R.; Sang, R.; Musila, L. Genetic divergence of Chikungunya virus plaque variants from the Comoros Island (2005). Virus Genes 2015, 51, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Sergon, K.; Yahaya, A.A.; Brown, J.; Bedja, S.A.; Mlindasse, M.; Agata, N.; Allaranger, Y.; Ball, M.D.; Powers, A.M.; Ofula, V.; et al. Seroprevalence of Chikungunya virus infection on Grande Comore Island, union of the Comoros, 2005. Am. J. Trop. Med. Hyg. 2007, 76, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Jaimipak, T.; Yoksan, S.; Ubol, S.; Pulmanausahakul, R. Small plaque size variant of Chikungunya primary isolate showed reduced virulence in mice. Asian Pac. J. Allergy Immunol. 2018, 36, 201–205. [Google Scholar] [PubMed]
- Abeyratne, E.; Freitas, J.R.; Zaid, A.; Mahalingam, S.; Taylor, A. Attenuation and stability of CHIKV-NoLS, a live-attenuated Chikungunya virus vaccine candidate. Vaccines 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Studer, E.; Michel, M. Two nucleolar targeting signals present in the N-terminal part of Semliki Forest virus capsid protein. Arch. Virol. 1994, 137, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Liu, X.; Zaid, A.; Goh, L.Y.H.; Hobson-Peters, J.; Hall, R.A.; Merits, A.; Mahalingam, S. Mutation of the N-terminal region of Chikungunya virus capsid protein: implications for vaccine design. MBio 2017, 8, e01970-16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Owen, K.E.; Choi, H.-K.; Lee, H.; Lu, G.; Wengler, G.; Brown, D.T.; Rossmann, M.G.; Kuhn, R.J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 1996, 4, 531–541. [Google Scholar] [CrossRef]
- Kuhn, J.; Becker, S.; Ebihara, H.; Geisbert, T.; Jahrling, P.; Kawaoka, Y.; Netesov, S.; Nichol, S.; Peters, C.; Volchkov, V. Family- Filoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 665–671. [Google Scholar]
- Bukreyev, A.; Rollin, P.E.; Tate, M.K.; Yang, L.; Zaki, S.R.; Shieh, W.-J.; Murphy, B.R.; Collins, P.L.; Sanchez, A. Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 2007, 81, 6379–6388. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-S.; Weng, T.-H.; Wu, X.-X.; Wang, F.X.C.; Lu, X.-Y.; Wu, H.-B.; Wu, N.-P.; Li, L.-J.; Yao, H.-P. The lifecycle of the Ebola virus in host cells. Oncotarget 2017, 8, 55750–55759. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Andersen, K.G.; Baize, S.; Bào, Y.; Bavari, S.; Berthet, N.; Blinkova, O.; Brister, J.R.; Clawson, A.N.; Fair, J.; et al. Nomenclature- and database-compatible names for the two Ebola virus variants that emerged in Guinea and the Democratic Republic of the Congo in 2014. Viruses 2014, 6, 4760–4799. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- World Health Organization. Ebola Virus Disease. Available online: https://www.who.int/en/news-room/fact-sheets/detail/ebola-virus-disease (accessed on 5 April 2019).
- Bell, B.P.; Damon, I.K.; Jernigan, D.B.; Kenyon, T.A.; Nichol, S.T.; O’Connor, J.P.; Tappero, J.W. Overview, control strategies and lessons learned in the CDC response to the 2014–2016 Ebola epidemic. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Hu, Y.; Liang, Q.; Wei, M.; Zhu, F. Ebola vaccines in clinical trial: The promising candidates. Hum. Vaccin. Immunother. 2016, 13, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.G.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Grifoni, A.; Lo Presti, A.; Cella, E.; Montesano, C.; Zehender, G.; Colizzi, V.; Amicosante, M.; Ciccozzi, M. Amino acid mutations in Ebola virus glycoprotein of the 2014 epidemic. J. Med. Virol. 2015, 87, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.T.; Kurosaki, Y.; Izumi, T.; Nakano, Y.; Oloniniyi, O.K.; Yasuda, J.; Koyanagi, Y.; Sato, K.; Nakagawa, S. Functional mutations in spike glycoprotein of Zaire Ebolavirus associated with an increase in infection efficiency. Genes Cells 2017, 22, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Diehl, W.E.; Lin, A.E.; Grubaugh, N.D.; Carvalho, L.M.; Kim, K.; Kyawe, P.P.; McCauley, S.M.; Donnard, E.; Kucukural, A.; McDonel, P. Ebola virus glycoprotein with increased infectivity dominated the 2013–2016 epidemic. Cell 2016, 167, 1088–1098. [Google Scholar] [CrossRef]
- Urbanowicz, R.A.; McClure, C.P.; Sakuntabhai, A.; Sall, A.A.; Kobinger, G.; Müller, M.A.; Holmes, E.C.; Rey, F.A.; Simon-Loriere, E.; Ball, J.K. Human adaptation of Ebola virus during the West African outbreak. Cell 2016, 167, 1079–1087. [Google Scholar] [CrossRef]
- Hoffmann, M.; Crone, L.; Dietzel, E.; Paijo, J.; González-Hernández, M.; Nehlmeier, I.; Kalinke, U.; Becker, S.; Pöhlmann, S. A polymorphism within the internal fusion loop of the Ebola virus glycoprotein modulates host cell entry. J. Virol. 2017, 91, e00177-17. [Google Scholar] [CrossRef]
- Wang, M.K.; Lim, S.-Y.; Lee, S.M.; Cunningham, J.M. Biochemical basis for increased activity of Ebola glycoprotein in the 2013–16 epidemic. Cell Host Microbe 2017, 21, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, E.; Schudt, G.; Krähling, V.; Matrosovich, M.; Becker, S. Functional characterization of adaptive mutations during the West African Ebola virus outbreak. J. Virol. 2017, 91, e01913-16. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, Y.; Ueda, M.T.; Nakano, Y.; Yasuda, J.; Koyanagi, Y.; Sato, K.; Nakagawa, S. Different effects of two mutations on the infectivity of Ebola virus glycoprotein in nine mammalian species. J. Gen. Virol. 2018, 99, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.W.; Matthews, D.A.; Hiscox, J.A.; Elmore, M.J.; Pollakis, G.; Rambaut, A.; Hewson, R.; García-Dorival, I.; Bore, J.A.; Koundouno, R.; et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 2015, 524, 97. [Google Scholar] [CrossRef] [PubMed]
- Fedewa, G.; Radoshitzky, S.R.; Chī, X.; Dǒngb, L.; Spear, M.; Strauli, N.; Stenglein, M.D.; Hernandez, R.D.; Jahrling, P.B.; Kuhn, J.H.; et al. Genomic characterization of serial-passaged Ebola virus in a boa constrictor cell line. bioRxiv 2016, 091603. [Google Scholar]
- de Groot, R.J.; Baker, S.; Baric, R.; Enjuanes, L.; Gorbalenya, A.; Holmes, K.; Perlman, S.; Poon, L.; Rottier, P.; Talbot, P. Family- Coronaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 806–828. [Google Scholar]
- Memish, Z.A.; Assiri, A.; Almasri, M.; Alhakeem, R.F.; Turkestani, A.; Al Rabeeah, A.A.; Al-Tawfiq, J.A.; Alzahrani, A.; Azhar, E.; Makhdoom, H.Q.; et al. Prevalence of MERS-CoV nasal carriage and compliance with the Saudi health recommendations among pilgrims attending the 2013 Hajj. J. Infect. Dis. 2014, 210, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, I.; Corman, V.M.; Müller, M.A.; Lenk, M.; Ulrich, R.G.; Drosten, C. Replicative capacity of MERS Coronavirus in livestock cell lines. Emerg. Infect. Dis. 2014, 20, 276–279. [Google Scholar] [CrossRef]
- Cetra, A. MERS-CoV update: What you need to know. Lancet Infect. Dis. 2014, 14, 924–925. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Severe Respiratory Disease Associated with Middle East Respiratory Syndrome Coronavirus (MERS-CoV), 22nd ed.; ECDC: Stockholm, Sweden, 2018. [Google Scholar]
- Terada, Y.; Kawachi, K.; Matsuura, Y.; Kamitani, W. MERS Coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 2017, 511, 95–105. [Google Scholar] [CrossRef]
- Lu, X.; Rowe, L.A.; Frace, M.; Stevens, J.; Abedi, G.R.; Elnile, O.; Banassir, T.; Al-Masri, M.; Watson, J.T.; Assiri, A.; et al. Spike gene deletion quasispecies in serum of patient with acute MERS-CoV infection. J. Med. Virol. 2017, 89, 542–545. [Google Scholar] [CrossRef]
- Ou, X.; Zheng, W.; Shan, Y.; Mu, Z.; Dominguez, S.R.; Holmes, K.V.; Qian, Z. Identification of the fusion peptide-containing region in Betacoronavirus spike glycoproteins. J. Virol. 2016, 90, 5586–5600. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Weber, H.; Elzayat, M.T.; Wang, L.; Graham, B.S.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J. Virol. 2019, 93, e01381-18. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Huh, H.J.; Kim, Y.J.; Son, D.-S.; Jeon, H.-J.; Im, E.-H.; Kim, J.-W.; Lee, N.Y.; Kang, E.-S.; Kang, C.I.; et al. Analysis of intrapatient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001214. [Google Scholar] [CrossRef] [PubMed]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Mishra, N.; Jain, K.; Zalmout, I.S.; Jabado, O.J.; Karesh, W.B.; Daszak, P.; Mohammed, O.B.; Alagaili, A.N.; Lipkin, W.I. Middle East respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. MBio 2014, 5, e01146-14. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: 2018b Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 18 July 2019).
- MacLachlan, N.J.; Dubovi, E.J. Chapter 17—Paramyxoviridae and Pneumoviridae. In Fenner’s Veterinary Virology, 5th ed.; MacLachlan, N.J., Dubovi, E.J., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 327–356. [Google Scholar]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Chapter 26—Paramyxoviruses. In Fenner and White’s Medical Virology, 5th ed.; Burrell, C.J., Howard, C.R., Murphy, F.A., Eds.; Academic Press: London, UK, 2017; pp. 367–382. [Google Scholar]
- Biesbroeck, L.; Sidbury, R. Viral exanthems: an update. Dermatol. Ther. 2013, 26, 433–438. [Google Scholar] [CrossRef]
- Ludlow, M.; McQuaid, S.; Milner, D.; de Swart, R.L.; Duprex, W.P. Pathological consequences of systemic measles virus infection. J. Pathol. 2015, 235, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Shirogane, Y.; Sato, Y.; Hashiguchi, T.; Yanagi, Y. New Insights into Measles Virus Brain Infections. Trends Microbiol. 2019, 27, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Huiming, Y.; Chaomin, W.; Meng, M. Vitamin A for treating measles in children. Cochrane Database Syst. Rev. 2005, 4, CD001479. [Google Scholar]
- Pfaller, C.K.; Cattaneo, R.; Schnell, M.J. Reverse genetics of Mononegavirales: How they work, new vaccines and new cancer therapeutics. Virology 2015, 479–480, 331–344. [Google Scholar] [CrossRef]
- Palosaari, H.; Parisien, J.-P.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 2003, 77, 7635–7644. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.; Hodge, G.; McChesney, M.B.; Cattaneo, R. Attenuation of V- or C-defective measles viruses: Infection control by the inflammatory and interferon responses of rhesus monkeys. J. Virol. 2008, 82, 5359–5367. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Takeda, M.; Ohno, S.; Shirogane, Y.; Iwasaki, M.; Yanagi, Y. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 2008, 82, 8296–8306. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Parisien, J.-P.; Horvath, C.M. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J. Virol. 2008, 82, 8330–8338. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Schmid, A.; Eschle, D.; Baczko, K.; ter Meulen, V.; Billeter, M.A. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 1988, 55, 255–265. [Google Scholar] [CrossRef]
- Paddock, M. What to Know About Mumps; University of Illinois-Chicago, School of Medicine: Chicago, IL, USA, 2017. [Google Scholar]
- Sauder, C.J.; Vandenburgh, K.M.; Iskow, R.C.; Malik, T.; Carbone, K.M.; Rubin, S.A. Changes in mumps virus neurovirulence phenotype associated with quasispecies heterogeneity. Virology 2006, 350, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Ganar, K.; Das, M.; Sinha, S.; Kumar, S. Newcastle disease virus: Current status and our understanding. Virus Res. 2014, 184, 71–81. [Google Scholar] [CrossRef]
- Miller, P.J.; Koch, G. Newcastle disease. In Diseases of Poultry, 13th ed.; Swayne, D.E., Ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2013; pp. 89–138. [Google Scholar]
- Alders, R.G. Making Newcastle disease vaccines available at village level. Vet. Rec. 2014, 174, 502–503. [Google Scholar] [CrossRef]
- Villar, E.; Barroso, I.M. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: A minireview. Glycoconj. J. 2006, 23, 5–17. [Google Scholar] [CrossRef]
- Meng, C.; Qiu, X.; Yu, S.; Li, C.; Sun, Y.; Chen, Z.; Liu, K.; Zhang, X.; Tan, L.; Song, C.; et al. Evolution of Newcastle Disease Virus Quasispecies Diversity and Enhanced Virulence after Passage through Chicken Air Sacs. J. Virol. 2015, 90, 2052–2063. [Google Scholar] [CrossRef]
- Gould, A.R.; Kattenbelt, J.A.; Selleck, P.; Hansson, E.; Della-Porta, A.; Westbury, H.A. Virulent Newcastle disease in Australia: Molecular epidemiological analysis of viruses isolated prior to and during the outbreaks of 1998–2000. Virus Res. 2001, 77, 51–60. [Google Scholar] [CrossRef]
- Kattenbelt, J.A.; Stevens, M.P.; Gould, A.R. Sequence variation in the Newcastle disease virus genome. Virus Res. 2006, 116, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Kattenbelt, J.A.; Stevens, M.P.; Selleck, P.W.; Gould, A.R. Analysis of Newcastle disease virus quasispecies and factors affecting the emergence of virulent virus. Arch. Virol. 2010, 155, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV virus taxonomy profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef] [PubMed]
- Bont, L.; Checchia, P.A.; Fauroux, B.; Figueras-Aloy, J.; Manzoni, P.; Paes, B.; Simões, E.A.F.; Carbonell-Estrany, X. Defining the epidemiology and burden of severe Respiratory Syncytial Virus infection among infants and children in western countries. Infect. Dis. Ther. 2016, 5, 271–298. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Respiratory Syncytial Virus Infection (RSV). Available online: https://www.cdc.gov/rsv/research/us-surveillance.html (accessed on 26 July 2019).
- Morrison, W.I.; Taylor, G.; Gaddum, R.M.; Ellis, S.A. Contribution of advances in immunology to vaccine development. Adv. Vet. Med. 1999, 41, 181–195. [Google Scholar] [PubMed]
- Zhao, X.; Chen, F.-P.; Sullender, W.M. Respiratory syncytial virus escape mutant derived in vitro resists palivizumab prophylaxis in cotton rats. Virology 2004, 318, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Deplanche, M.; Lemaire, M.; Mirandette, C.; Bonnet, M.; Schelcher, F.; Meyer, G. In vivo evidence for quasispecies distributions in the bovine respiratory syncytial virus genome. J. Gen. Virol. 2007, 88, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.E.; Tjørnehøj, K.; Viuff, B. Extensive Sequence Divergence among Bovine Respiratory Syncytial Viruses Isolated during Recurrent Outbreaks in Closed Herds. J. Clin. Microbiol. 2000, 38, 4222. [Google Scholar]
- Wellehan, J.; Cortes-Hinojosa, G. 84—Marine Mammal Viruses. In Fowler’s Zoo and Wild Animal Medicine Current Therapy; Miller, R.E., Lamberski, N., Calle, P.P., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2019; Volume 9, pp. 597–602. [Google Scholar]
- McCauley, J.W.; Hongo, S.; Kaverin, N.V.; Kochs, G.; Lamb, R.A.; Matrosovich, M.N.; Perez, D.R.; Palese, P.; Presti, R.M.; Rimstad, E.; et al. Family-Orthomyxoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 749–761. [Google Scholar]
- World Health Organization. Seasonal Influenza. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 20 July 2019).
- Zost, S.J.; Parkhouse, K.; Gumina, M.E.; Kim, K.; Perez, S.D.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 Influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef]
- Simon, B.; Pichon, M.; Valette, M.; Burfin, G.; Richard, M.; Lina, B.; Josset, L. Whole genome sequencing of a (H3N2) Influenza viruses reveals variants associated with severity during the 2016–2017 season. Viruses 2019, 11, 108. [Google Scholar] [CrossRef]
- Dietze, K.; Graaf, A.; Homeier-Bachmann, T.; Grund, C.; Forth, L.; Pohlmann, A.; Jeske, C.; Wintermann, M.; Beer, M.; Conraths, F.J. From low to high pathogenicity—Characterization of H7N7 Avian Influenza viruses in two epidemiologically linked outbreaks. Transbound. Emerg. Dis. 2018, 65, 1576–1587. [Google Scholar] [CrossRef]
- Watanabe, Y.; Arai, Y.; Kawashita, N.; Ibrahim, M.S.; Elgendy, E.M.; Daidoji, T.; Kajikawa, J.; Hiramatsu, H.; Sriwilaijaroen, N.; Ono, T. Characterization of H5N1 Influenza virus quasispecies with adaptive hemagglutinin mutations from single-virus infections of human airway cells. J. Virol. 2018, 92, e02004-17. [Google Scholar] [CrossRef] [PubMed]
- Barbezange, C.; Jones, L.; Blanc, H.; Isakov, O.; Celniker, G.; Enouf, V.; Shomron, N.; Vignuzzi, M.; van der Werf, S. Seasonal genetic drift of human influenza A virus quasispecies revealed by deep sequencing. Front. Microbiol. 2018, 9, 2596. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, H.; Seidel, N.; Blaess, M.F.; Claus, R.A.; Linde, J.; Slevogt, H.; Sauerbrei, A.; Guthke, R.; Schmidtke, M. Differential biphasic transcriptional host response associated with coevolution of hemagglutinin quasispecies of Influenza A virus. Front. Microbiol. 2016, 7, 1167. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Sun, H.; Sun, Z.; Sun, Y.; Kong, W.; Pu, J.; Ma, G.; Yin, Y.; Yang, H.; Guo, X. Influenza A virus acquires enhanced pathogenicity and transmissibility after serial passages in swine. J. Virol. 2014, 88, 11981–11994. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Hepadnavirus genome replication and persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus taxonomy and Hepatitis B virus genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef]
- You, C.R.; Lee, S.W.; Jang, J.W.; Yoon, S.K. Update on Hepatitis B virus infection. World J. Gastroenterol. 2014, 20, 13293. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and genetic variability of Hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Revill, P.A.; Penicaud, C.; Brechot, C.; Zoulim, F. Meeting the challenge of eliminating chronic Hepatitis B infection. Genes 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Homs, M.; Buti, M.; Tabernero, D.; Quer, J.; Sanchez, A.; Corral, N.; Esteban, R.; Rodriguez-Frias, F. Quasispecies dynamics in main core epitopes of Hepatitis B virus by ultra-deep-pyrosequencing. World J. Gastroenterol. 2012, 18, 6096. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, S.; Zhang, Y.; Zhang, M.; Lv, L.; Huang, C.; Li, X.; Li, J.; Zhang, Z. Viral quasispecies of Hepatitis B virus in patients with YMDD mutation and lamivudine resistance may not predict the efficacy of lamivudine/adefovir rescue therapy. Exp. Ther. Med. 2019, 17, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-Y.; Liu, C.; Chien, W.-H.; Wu, L.-L.; Tao, Y.; Wu, D.; Lu, X.; Hsieh, C.-H.; Chen, P.-J.; Wang, H.-Y. New insights into the evolutionary rate of Hepatitis B virus at different biological scales. J. Virol. 2015, 89, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Forbi, J.C.; Vaughan, G.; Purdy, M.A.; Campo, D.S.; Xia, G.-l.; Ganova-Raeva, L.M.; Ramachandran, S.; Thai, H.; Khudyakov, Y.E. Epidemic history and evolutionary dynamics of Hepatitis B virus infection in two remote communities in rural Nigeria. PLoS ONE 2010, 5, e11615. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandary, M.B.; Masomian, M.; Poh, C.L. Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. Int. J. Mol. Sci. 2019, 20, 4657. https://doi.org/10.3390/ijms20184657
Mandary MB, Masomian M, Poh CL. Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. International Journal of Molecular Sciences. 2019; 20(18):4657. https://doi.org/10.3390/ijms20184657
Chicago/Turabian StyleMandary, Madiiha Bibi, Malihe Masomian, and Chit Laa Poh. 2019. "Impact of RNA Virus Evolution on Quasispecies Formation and Virulence" International Journal of Molecular Sciences 20, no. 18: 4657. https://doi.org/10.3390/ijms20184657
APA StyleMandary, M. B., Masomian, M., & Poh, C. L. (2019). Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. International Journal of Molecular Sciences, 20(18), 4657. https://doi.org/10.3390/ijms20184657