A Multi-Layered Study on Harmonic Oscillations in Mammalian Genomics and Proteomics



 ,
,  , and
, and 
Abstract
1. Introduction
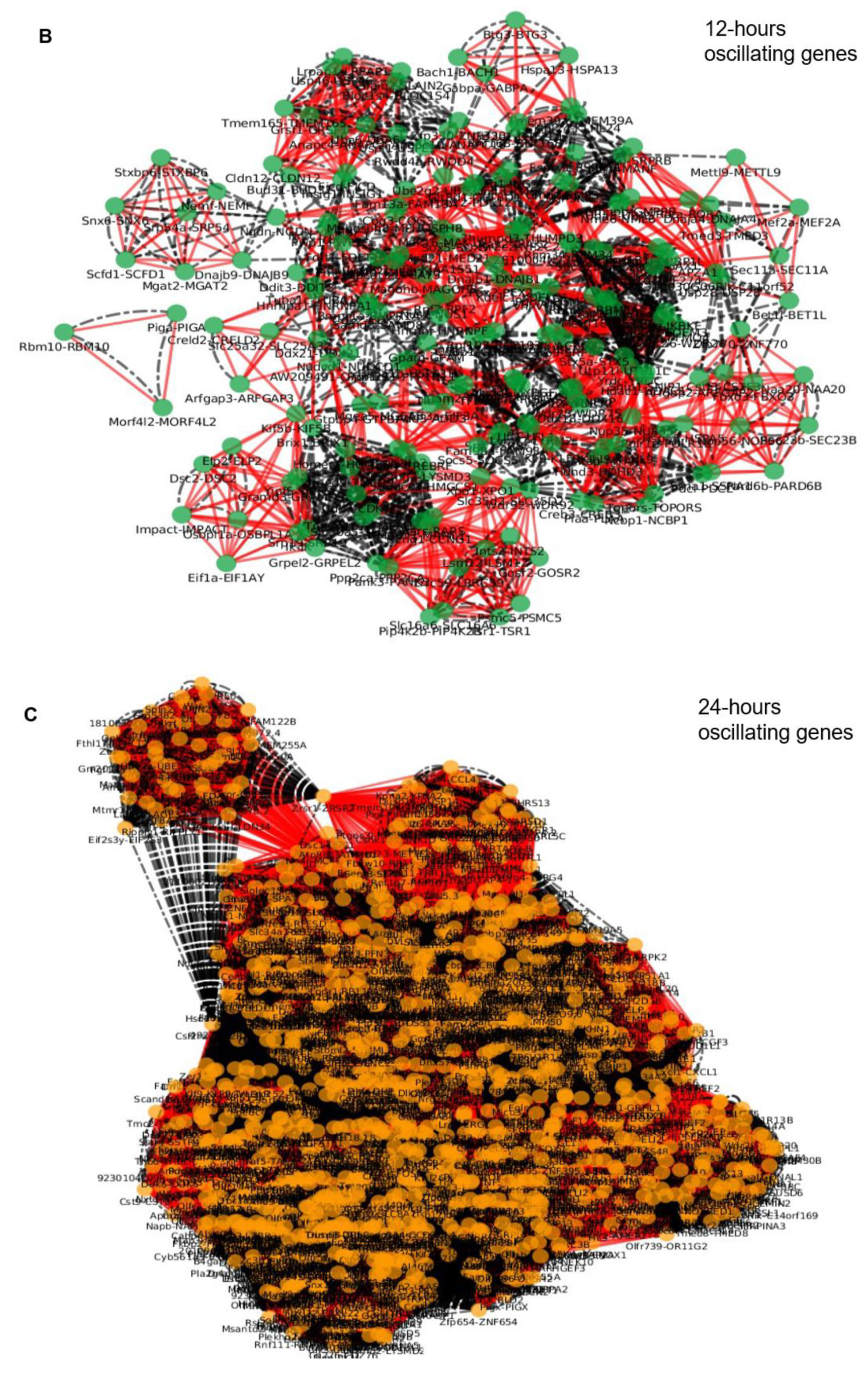
2. Results
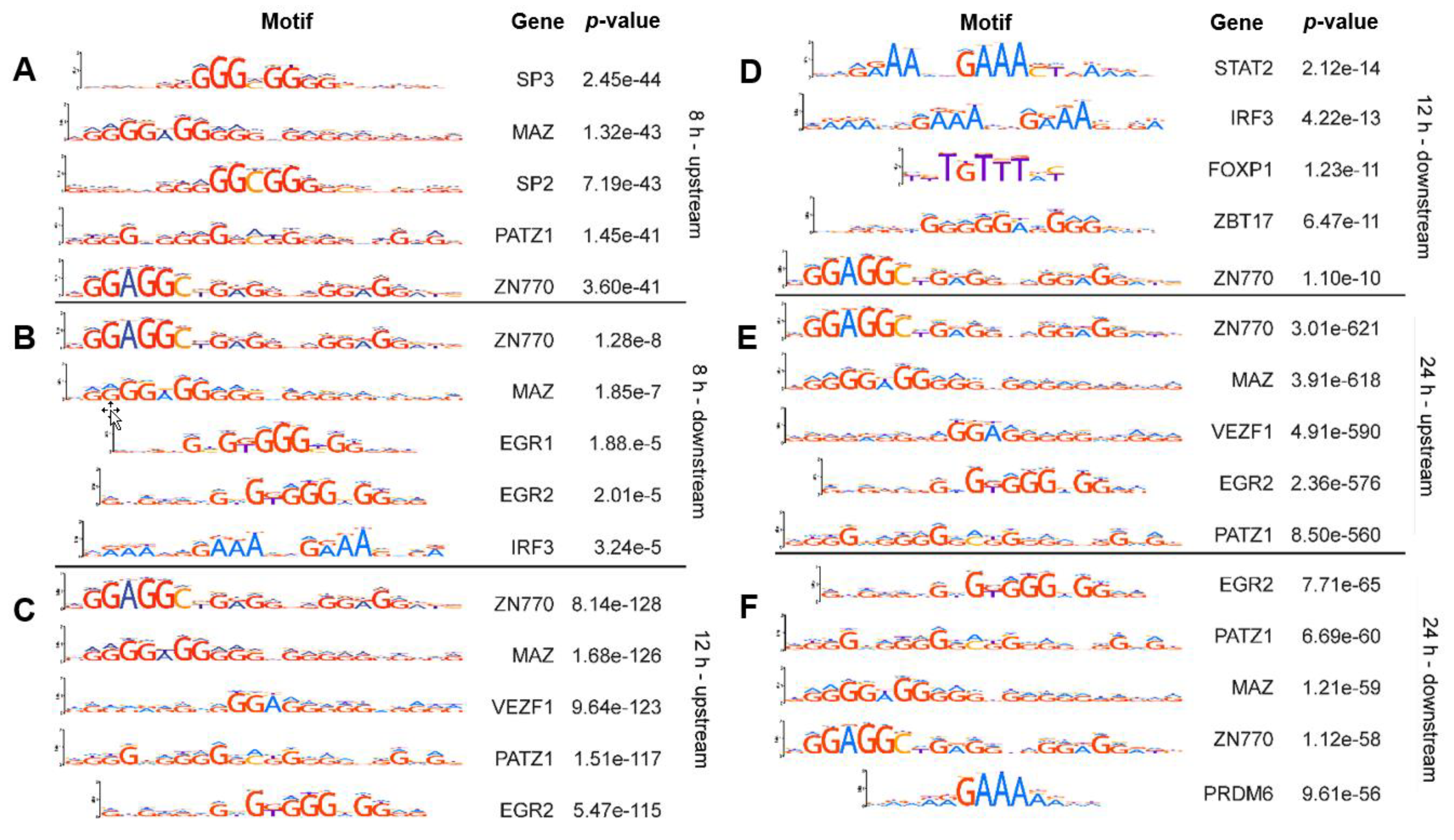
2.1. Known Transcription Factor Binding Sites Are Enriched in the Promoter Regions of the 8-h, 12-h, and 24-h Rhytmically Expressed Genes
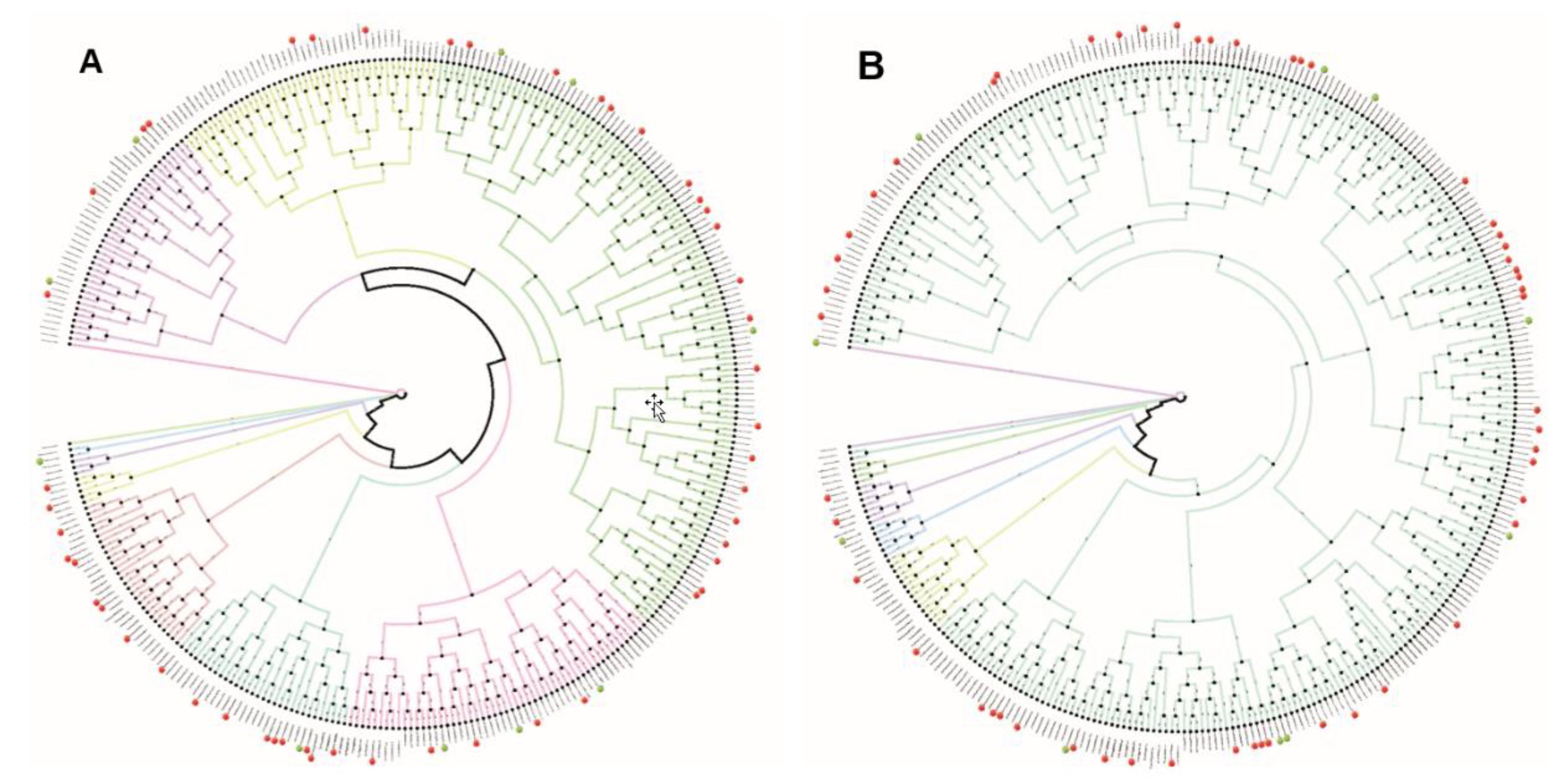
2.2. Phylogenetic Analysis Shows Similarity within the Promoter Regions of the 8 h and 12 h Gene Sets
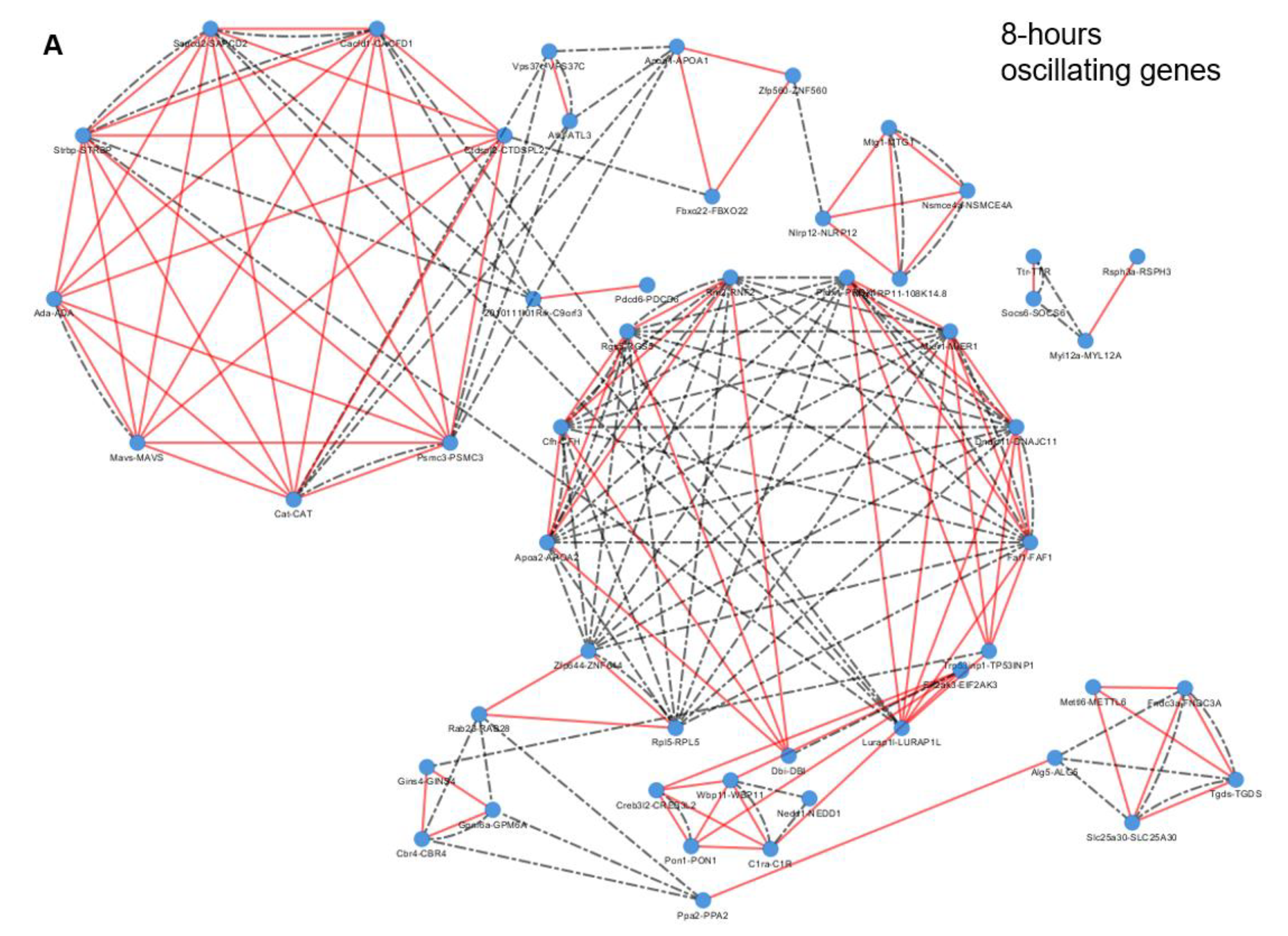
2.3. Epigenetic and Non-epigenetic Regulation of Oscillating Gene Expression
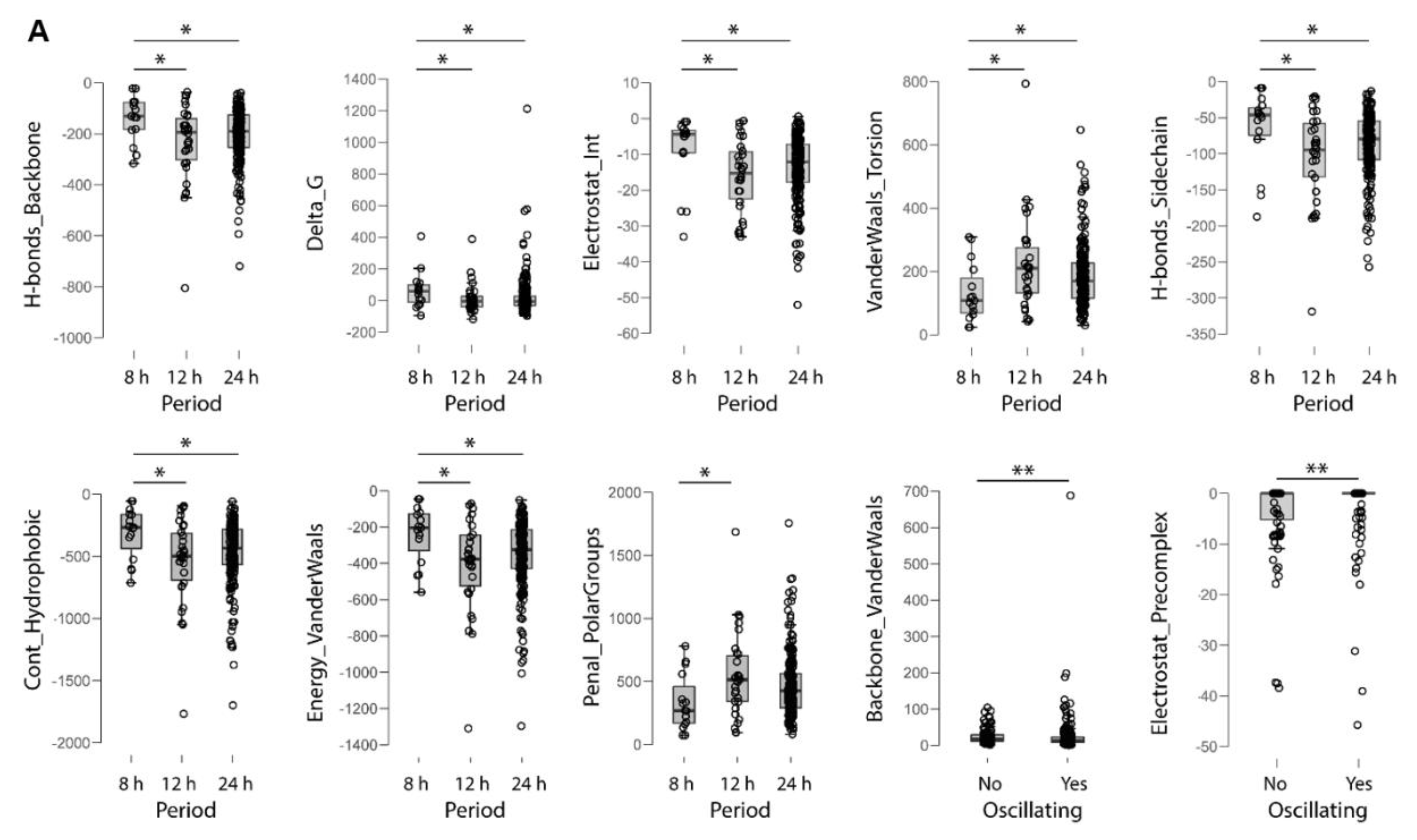
2.4. Electrochemical Properties of Oscillating Proteins
2.5. Chromosome Mapping of Oscillating Genes
3. Discussion
3.1. Oscillating Proteins Are Hallmarked by Higher Overall Stability when Compared to Non-Oscillating Proteins
3.2. Specific Enriched Processes Govern the Regulation and Output of the Ultradian Oscillating Genes
3.3. Homology Mapping of Oscillating Genes Revealed Different Degree of Localization Conservation for the Three Gene Sets
4. Materials and Methods
4.1. Primary Dataset
4.2. Sequence Analysis for Known Transcription Factor Binding Sites
4.3. Multiple Sequence Alignment and Phylogenetic Analysis
4.4. Enrichment Analysis of the 8-h, 12-h and 24-h Gene Sets
4.5. Randomized Network Analysis on Ultradian and Circadian Genes
4.6. Epigenetic and Non-epigenetic Regulation of Oscillating Gene Expression
4.7. Impact of Protein Expression on Survival
4.8. Electrochemical Properties of Oscillating Proteins
4.9. Chromosome Mapping of Oscillating Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, J.C. Molecular bases for circadian clocks. Cell 1999, 96, 271–290. [Google Scholar] [CrossRef]
- Mermet, J.; Yeung, J.; Naef, F. Systems Chronobiology: Global Analysis of Gene Regulation in a 24-Hour Periodic World. Cold Spring Harb. Perspect. Biol. 2017, 9, a028720. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.; Saini, C.; Curie, T.; Emmenegger, Y.; Rando, G.; Gosselin, P.; Gotic, I.; Gos, P.; Franken, P.; Schibler, U. The systemic control of circadian gene expression. Diabetes Obes. Metab. 2015, 17 (Suppl. S1), 23–32. [Google Scholar] [CrossRef]
- Bozek, K.; Relogio, A.; Kielbasa, S.M.; Heine, M.; Dame, C.; Kramer, A.; Herzel, H. Regulation of clock-controlled genes in mammals. PLoS ONE 2009, 4, e4882. [Google Scholar] [CrossRef]
- Lowrey, P.L.; Takahashi, J.S. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 2011, 74, 175–230. [Google Scholar]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef]
- Lehmann, R.; Childs, L.; Thomas, P.; Abreu, M.; Fuhr, L.; Herzel, H.; Leser, U.; Relogio, A. Assembly of a comprehensive regulatory network for the mammalian circadian clock: A bioinformatics approach. PLoS ONE 2015, 10, e0126283. [Google Scholar] [CrossRef]
- Hirano, A.; Fu, Y.H.; Ptacek, L.J. The intricate dance of post-translational modifications in the rhythm of life. Nat. Struct. Mol. Biol. 2016, 23, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Selby, C.P.; Chiou, Y.Y.; Ozkan-Dagliyan, I.; Gaddameedhi, S.; Sancar, A. Dual modes of CLOCK: BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 2014, 28, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Aryal, R.P.; Kwak, P.B.; Tamayo, A.G.; Gebert, M.; Chiu, P.L.; Walz, T.; Weitz, C.J. Macromolecular Assemblies of the Mammalian Circadian Clock. Mol. Cell 2017, 67, 770–782 e6. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.E.; DiTacchio, L.; Hayes, K.R.; Vollmers, C.; Pulivarthy, S.; Baggs, J.E.; Panda, S.; Hogenesch, J.B. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009, 5, e1000442. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Nagoshi, E.; Brown, S.A.; Ripperger, J.; Schibler, U. The mammalian circadian timing system: From gene expression to physiology. Chromosoma 2004, 113, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, L.; Abreu, M.; Pett, P.; Relogio, A. Circadian systems biology: When time matters. Comput. Struct. Biotechnol. J. 2015, 13, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.O.; Herzel, H. Mechanism for 12 hr rhythm generation by the circadian clock. Cell Rep. 2013, 3, 1228–1238. [Google Scholar] [CrossRef]
- Martins, B.M.; Das, A.K.; Antunes, L.; Locke, J.C. Frequency doubling in the cyanobacterial circadian clock. Mol. Syst. Biol. 2016, 12, 896. [Google Scholar] [CrossRef]
- Shinohara, Y.; Koyama, Y.M.; Ukai-Tadenuma, M.; Hirokawa, T.; Kikuchi, M.; Yamada, R.G.; Ukai, H.; Fujishima, H.; Umehara, T.; Tainaka, K.; et al. Temperature-Sensitive Substrate and Product Binding Underlie Temperature-Compensated Phosphorylation in the Clock. Mol. Cell 2017, 67, 783–798 e20. [Google Scholar] [CrossRef]
- Isojima, Y.; Nakajima, M.; Ukai, H.; Fujishima, H.; Yamada, R.G.; Masumoto, K.H.; Kiuchi, R.; Ishida, M.; Ukai-Tadenuma, M.; Minami, Y.; et al. CKIepsilon/delta-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 2009, 106, 15744–15749. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Mazza, T.; Capocefalo, D.; Genov, N.; Biagini, T.; Fusilli, C.; Scholkmann, F.; Relogio, A.; Hogenesch, J.B.; Mazzoccoli, G. Systematic Analysis of Mouse Genome Reveals Distinct Evolutionary and Functional Properties Among Circadian and Ultradian Genes. Front. Physiol. 2018, 9, 1178. [Google Scholar] [CrossRef] [PubMed]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M. Trends in substitution models of molecular evolution. Front. Genet. 2015, 6, 319. [Google Scholar] [CrossRef] [PubMed]
- El-Athman, R.; Fuhr, L.; Relogio, A. A Systems-Level Analysis Reveals Circadian Regulation of Splicing in Colorectal Cancer. EBioMedicine 2018, 33, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Genov, N.; Basti, A.; Abreu, M.; Astaburuaga, R.; Relogio, A. A bioinformatic analysis identifies circadian expression of splicing factors and time-dependent alternative splicing events in the HD-MY-Z cell line. Sci. Rep. 2019, 9, 11062. [Google Scholar] [CrossRef] [PubMed]
- Genov, N.; Basti, A.; Abreu, M.; Relogio, A. Temporal Splicing Switches in Elements of the TNF-Pathway Identified by Computational Analysis of Transcriptome Data for Human Cell Lines. Int. J. Mol. Sci. 2019, 20, 1182. [Google Scholar] [CrossRef]
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, e2002940. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–97. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhang, Q.; Pan, Y.; Mace, E.M.; York, B.; Antoulas, A.C.; Dacso, C.C.; O’Malley, B.W. A Cell-Autonomous Mammalian 12 hr Clock Coordinates Metabolic and Stress Rhythms. Cell Metab. 2017, 25, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Rivas, G.; Minton, A.P. Macromolecular Crowding In Vitro, In Vivo, and In Between. Trends Biochem. Sci. 2016, 41, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–208. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Medvedeva, Y.A.; Schaefer, U.; Kasianov, A.S.; Vorontsov, I.E.; Bajic, V.B.; Makeev, V.J. HOCOMOCO: A comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013, 41, D195–202. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Oscillating Proteins | Non-Oscillating Proteins | p | Period 8 h | Period 12 h | Period 24 h | p-value |
|---|---|---|---|---|---|---|---|
| Residue Number | 323.7 ± 193.6 | 313.3 ± 165.5 | 0.642 | 224.0 ± 136.6 | 373.3 ± 232.1 | 321.8 ± 188.5 | 0.044* (8 h vs. 12 h) 0.063 (8 h vs. 24 h) 0.347 (12 vs. 24 h) |
| Delta_G | 23.34 ± 123.04 | 29.84 ± 91.09 | 0.065 | 81.91 ± 133.64 | 13.53 ± 95.47 | 21.64 ± 125.74 | 0.047* (8 h vs. 12 h) 0.047* (8 h vs. 24 h) 0.377 (12 vs. 24 h) |
| H-bonds_Backbone | −206.6 ± 112.7 | −209.4 ± 121.7 | 0.429 | −135.4 ± 95.49 | −236.2 ± 157.46 | −206 ± 104.26 | 0.022* (8 h vs. 12 h) 0.022* (8 h vs. 24 h) 0.221 (12 vs. 24 h) |
| H-bonds_Sidechain | −88.50 ± 49.24 | −89.31 ± 52.65 | 0.496 | −61.66 ± 56.19 | −103.98 ± 65.35 | −87.67 ± 45.53 | 0.009** (8 h vs. 12 h) 0.016* (8 h vs. 24 h) 0.125 (12 vs. 24 h) |
| Energy_VanderWaals | −355.8 ± 201.6 | −363.0 ± 211.2 | 0.465 | −239.1 ± 168.5 | −407.4 ± 260.7 | −354.5 ± 191.4 | 0.033* (8 h vs. 12 h) 0.043* (8 h vs. 24 h) 0.175 (12 vs. 24 h) |
| Electrostat_Int | −13.75 ± 9.226 | −14.03 ± 10.912 | 0.395 | −8.291 ± 9.198 | −16.011 ± 9.858 | −13.710 ± 9.049 | 0.006** (8 h vs. 12 h) 0.014* (8 h vs. 24 h) 0.091 (12 vs. 24 h) |
| Penal_PolarGroups | 474.9 ± 267.9 | 486.5 ± 278.3 | 0.401 | 335.0 ± 232.3 | 541.5 ± 339.8 | 472.6 ± 255.7 | 0.049* (8 h vs. 12 h) 0.071 (8 h vs. 24 h) 0.162 (12 vs. 24 h) |
| Cont_Hydrophobic | −472.4 ± 268.4 | −480.7 ± 281.7 | 0.470 | −311.7 ± 219.0 | −542.3 ± 350.6 | −470.8 ± 254.0 | 0.029* (8 h vs. 12 h) 0.037* (8 h vs. 24 h) 0.180 (12 vs. 24 h) |
| Penal_VanderWaals | 23.16 ± 49.59 | 24.85 ± 21.93 | 0.002* | 19.94 ± 27.56 | 22.23 ± 23.03 | 23.46 ± 53.31 | 0.444 (8 h vs. 12 h) 0.444 (8 h vs. 24 h) 0.444 (12 vs. 24 h) |
| VanderWaals_Torsion | 198.7 ± 108.4 | 191.1 ± 109.7 | 0.496 | 126.9 ± 91.05 | 223.1 ± 151.23 | 188.2 ± 100.39 | 0.023* (8 h vs. 12 h) 0.038* (8 h vs. 24 h) 0.140 (12 vs. 24 h) |
| Backbone_VanderWaals | 470.6 ± 272.1 | 485.1 ± 258.7 | 0.252 | 352.1 ± 228.7 | 532.7 ± 331.3 | 467.9 ± 263.5 | 0.081 (8 h vs. 12 h) 0.130 (8 h vs. 24 h) 0.141 (12 vs. 24 h) |
| Water Bonds | 0.699 ± 1.158 | 0.693 ± 1.024 | 0.458 | 0.237 ± 0.451 | 0.550 ± 0.742 | 0.745 ± 1.227 | 0.343 (8 h vs. 12 h) 0.259 (8 h vs. 24 h) 0.343 (12 vs. 24 h) |
| Electrostatic_HelixDipole | 11.66 ± 11.484 | 12.04 ± 8.639 | 0.095 | 8.414 ± 8.079 | 12.091 ± 9.241 | 11.769 ± 11.938 | 0.185 (8 h vs. 12 h) 0.185 (8 h vs. 24 h) 0.284 (12 vs. 24 h) |
| Cost_PeptideBond | −5.348 ± 4.489 | −5.511 ± 6.128 | 0.183 | −3.399 ± 3.684 | −5.705 ± 5.276 | −5.401 ± 4.399 | 0.131 (8 h vs. 12 h) 0.126 (8 h vs. 24 h) 0.496 (12 vs. 24 h) |
| Electrostat_Precomplex | −1.143 ± 4.972 | −3.606 ± 7.590 | <0.001* | 0.000 ± 0.000 | 0.000 ± 0.000 | −1.373 ± 5.422 | 0.500 (8 h vs. 12 h) 0.213 (8 h vs. 24 h) 0.073 (12 vs. 24 h) |
| Interaction_BoundMetals | −0.016 ± 0.155 | 0.000 ± 0.000 | 0.200 | 0.000 ± 0.000 | −0.053 ± 0.288 | −0.011 ± 0.129 | 0.594 (8 h vs. 12 h) 0.594 (8 h vs. 24 h) 0.594 (12 vs. 24 h) |
| Energy_Ionisation | −4.988 ± 12.64 | −6.075 ± 15.55 | 0.473 | −1.984 ± 4.856 | −8.547 ± 14.491 | −4.628 ± 12.589 | 0.444 (8 h vs. 12 h) 0.444 (8 h vs. 24 h) 0.444 (12 vs. 24 h) |
| Entropy_Complex | 1.220 ± 0.957 | 1.108 ± 0.820 | 0.226 | 0.865 ± 0.876 | 1.379 ± 1.0124 | 1.215 ± 0.951 | 0.134 (8 h vs. 12 h) 0.146 (8 h vs. 24 h) 0.223 (12 vs. 24 h) |
| M. musculus Network Specifics (after Isolate Nodes Removal) | |||
| Network | Nodes | Edges | Components |
| 8 h | 51 | 89 | 14 |
| 12 h | 199 | 1001 | 20 |
| 24 h | 1827 | 105970 | 20 |
| H. sapiens Network Specifics (after Isolate Nodes Removal) | |||
| 8 h | 49 | 92 | 14 |
| 12 h | 198 | 1085 | 23 |
| 24 h | 1826 | 95657 | 23 |
| Intersection Networks Characteristics | |||
| 8 h | 33 | 29 | 13 |
| 12 h | 45 | 169 | 312 |
| 24 h | 103 | 1801 | 33439 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genov, N.; Castellana, S.; Scholkmann, F.; Capocefalo, D.; Truglio, M.; Rosati, J.; Turco, E.M.; Biagini, T.; Carbone, A.; Mazza, T.; et al. A Multi-Layered Study on Harmonic Oscillations in Mammalian Genomics and Proteomics. Int. J. Mol. Sci. 2019, 20, 4585. https://doi.org/10.3390/ijms20184585
Genov N, Castellana S, Scholkmann F, Capocefalo D, Truglio M, Rosati J, Turco EM, Biagini T, Carbone A, Mazza T, et al. A Multi-Layered Study on Harmonic Oscillations in Mammalian Genomics and Proteomics. International Journal of Molecular Sciences. 2019; 20(18):4585. https://doi.org/10.3390/ijms20184585
Chicago/Turabian StyleGenov, Nikolai, Stefano Castellana, Felix Scholkmann, Daniele Capocefalo, Mauro Truglio, Jessica Rosati, Elisa Maria Turco, Tommaso Biagini, Annalucia Carbone, Tommaso Mazza, and et al. 2019. "A Multi-Layered Study on Harmonic Oscillations in Mammalian Genomics and Proteomics" International Journal of Molecular Sciences 20, no. 18: 4585. https://doi.org/10.3390/ijms20184585
APA StyleGenov, N., Castellana, S., Scholkmann, F., Capocefalo, D., Truglio, M., Rosati, J., Turco, E. M., Biagini, T., Carbone, A., Mazza, T., Relógio, A., & Mazzoccoli, G. (2019). A Multi-Layered Study on Harmonic Oscillations in Mammalian Genomics and Proteomics. International Journal of Molecular Sciences, 20(18), 4585. https://doi.org/10.3390/ijms20184585