Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications


Abstract
1. Introduction
2. DNA Methylation Enzymes in Hematopoiesis
3. Aberrant DNA Methylation in AML
3.1. Acute Promyelocytic Leukemia (APL) with PML-RARα
3.2. AML with MLL Gene Rearrangement
3.3. AML with DNMT3A Mutations
3.4. AML with TET2 Mutations
4. Clinical Implications of DNA Methylation in AML
4.1. Diagnostic Value of DNA Methylation
4.2. DNA Methylation in the Prognosis of AML
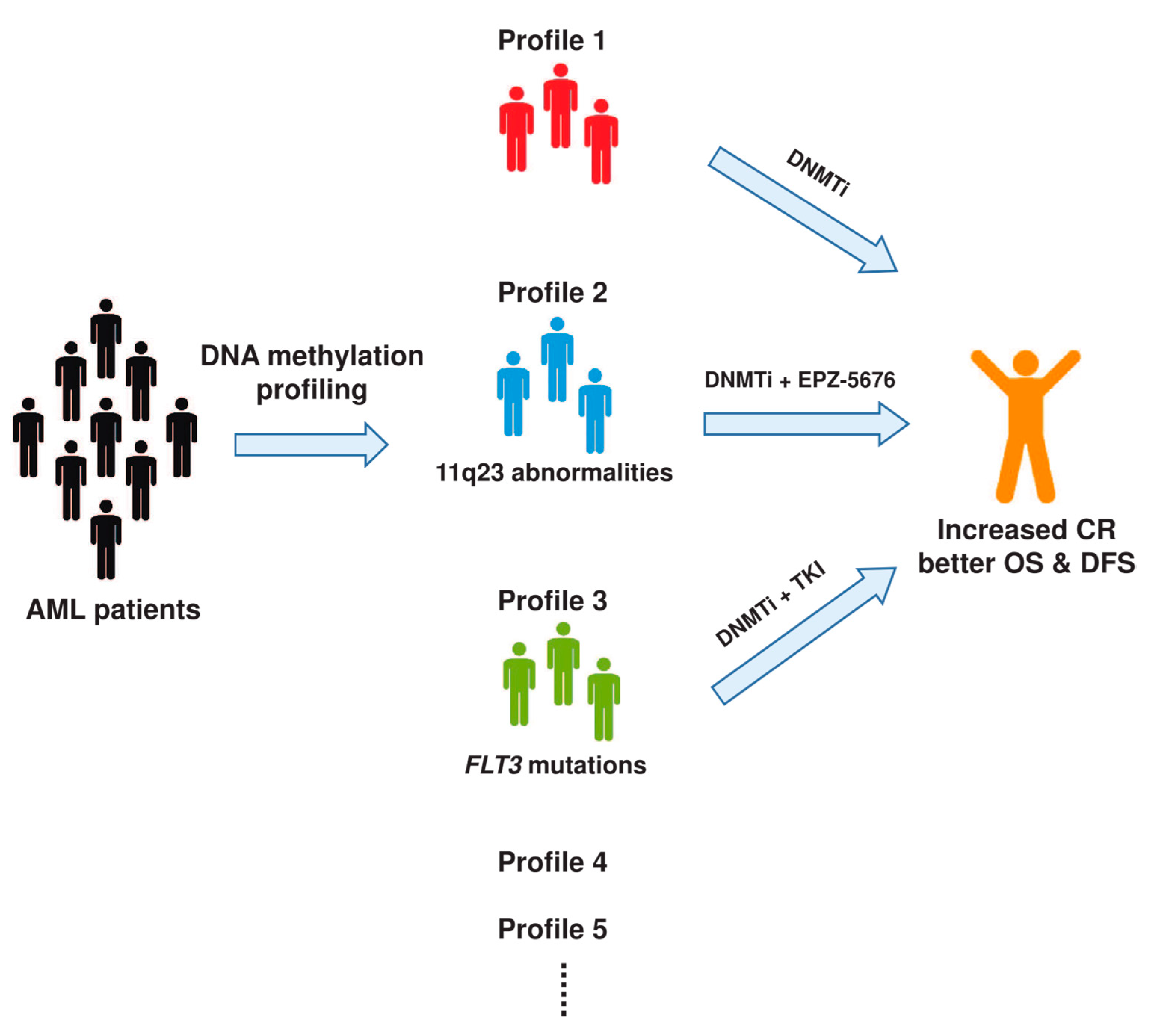
5. Targeting DNA Methylation in AML
6. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Cullen, S.M.; Mayle, A.; Rossi, L.; Goodell, M.A. Hematopoietic stem cell development: An epigenetic journey. Curr. Top. Dev. Biol. 2014, 107, 39–75. [Google Scholar] [PubMed]
- Kim, A.D.; Stachura, D.L.; Traver, D. Cell signaling pathways involved in hematopoietic stem cell specification. Exp. Cell Res. 2014, 329, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Link, D.C. The hematopoietic stem cell niche in homeostasis and disease. Blood 2015, 126, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Sashida, G.; Iwama, A. Epigenetic regulation of hematopoiesis. Int. J. Hematol. 2012, 96, 405–412. [Google Scholar] [CrossRef]
- Beerman, I.; Rossi, D.J. Epigenetic regulation of hematopoietic stem cell aging. Exp. Cell Res. 2014, 329, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R. DNA methylation and imprinting: Why bother? Trends Genet. 1997, 13, 323–329. [Google Scholar] [CrossRef]
- Schaefer, C.B.; Ooi, S.K.; Bestor, T.H.; Bourc’his, D. Epigenetic decisions in mammalian germ cells. Science 2007, 316, 398–399. [Google Scholar] [CrossRef]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Farlik, M.; Halbritter, F.; Muller, F.; Choudry, F.A.; Ebert, P.; Klughammer, J.; Farrow, S.; Santoro, A.; Ciaurro, V.; Mathur, A.; et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 2016, 19, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Broske, A.M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, Y.; Ema, H.; Okano, M.; Li, E.; Nakauchi, H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 2007, 204, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Schoofs, T.; Berdel, W.E.; Muller-Tidow, C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 2014, 28, 1–14. [Google Scholar] [CrossRef]
- Bock, C.; Halbritter, F.; Carmona, F.J.; Tierling, S.; Datlinger, P.; Assenov, Y.; Berdasco, M.; Bergmann, A.K.; Booher, K.; Busato, F.; et al. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726–737. [Google Scholar]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef]
- Luskin, M.R.; Gimotty, P.A.; Smith, C.; Loren, A.W.; Figueroa, M.E.; Harrison, J.; Sun, Z.; Tallman, M.S.; Paietta, E.M.; Litzow, M.R.; et al. A clinical measure of DNA methylation predicts outcome in de novo acute myeloid leukemia. JCI Insight 2016, 1, e87323. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef]
- Hogart, A.; Lichtenberg, J.; Ajay, S.S.; Anderson, S.; Center, N.I.H.I.S.; Margulies, E.H.; Bodine, D.M. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal overrepresentation of ETS transcription factor binding sites. Genome Res. 2012, 22, 1407–1418. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Pastor, W.A.; Koralov, S.B.; Rajewsky, K.; Rao, A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015, 263, 6–21. [Google Scholar] [CrossRef]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Pan, F.; Wingo, T.S.; Zhao, Z.; Gao, R.; Makishima, H.; Qu, G.; Lin, L.; Yu, M.; Ortega, J.R.; Wang, J.; et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat. Commun. 2017, 8, 15102. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, L.; Dawlaty, M.M.; Pan, F.; Weeks, O.; Zhou, Y.; Cao, Z.; Shi, H.; Wang, J.; Lin, L.; et al. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep. 2015, 13, 1692–1704. [Google Scholar] [CrossRef]
- An, J.; Gonzalez-Avalos, E.; Chawla, A.; Jeong, M.; Lopez-Moyado, I.F.; Li, W.; Goodell, M.A.; Chavez, L.; Ko, M.; Rao, A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015, 6, 10071. [Google Scholar] [CrossRef]
- Rousselot, P.; Hardas, B.; Patel, A.; Guidez, F.; Gaken, J.; Castaigne, S.; Dejean, A.; de The, H.; Degos, L.; Farzaneh, F.; et al. The PML-RAR alpha gene product of the t(15;17) translocation inhibits retinoic acid-induced granulocytic differentiation and mediated transactivation in human myeloid cells. Oncogene 1994, 9, 545–551. [Google Scholar]
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alcalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431. [Google Scholar] [CrossRef]
- Cole, C.B.; Verdoni, A.M.; Ketkar, S.; Leight, E.R.; Russler-Germain, D.A.; Lamprecht, T.L.; Demeter, R.T.; Magrini, V.; Ley, T.J. PML-RARA requires DNA methyltransferase 3A to initiate acute promyelocytic leukemia. J. Clin. Investig. 2016, 126, 85–98. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Villa, R.; Morey, L.; Raker, V.A.; Buschbeck, M.; Gutierrez, A.; De Santis, F.; Corsaro, M.; Varas, F.; Bossi, D.; Minucci, S.; et al. The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc. Natl. Acad. Sci. USA 2006, 103, 1400–1405. [Google Scholar] [CrossRef]
- Schoofs, T.; Rohde, C.; Hebestreit, K.; Klein, H.U.; Gollner, S.; Schulze, I.; Lerdrup, M.; Dietrich, N.; Agrawal-Singh, S.; Witten, A.; et al. DNA methylation changes are a late event in acute promyelocytic leukemia and coincide with loss of transcription factor binding. Blood 2013, 121, 178–187. [Google Scholar] [CrossRef]
- Martens, J.H.; Brinkman, A.B.; Simmer, F.; Francoijs, K.J.; Nebbioso, A.; Ferrara, F.; Altucci, L.; Stunnenberg, H.G. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 2010, 17, 173–185. [Google Scholar] [CrossRef]
- Saeed, S.; Logie, C.; Francoijs, K.J.; Frige, G.; Romanenghi, M.; Nielsen, F.G.; Raats, L.; Shahhoseini, M.; Huynen, M.; Altucci, L.; et al. Chromatin accessibility, p300, and histone acetylation define PML-RARalpha and AML1-ETO binding sites in acute myeloid leukemia. Blood 2012, 120, 3058–3068. [Google Scholar] [CrossRef]
- Alvarez, S.; Suela, J.; Valencia, A.; Fernandez, A.; Wunderlich, M.; Agirre, X.; Prosper, F.; Martin-Subero, J.I.; Maiques, A.; Acquadro, F.; et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS ONE 2010, 5, e12197. [Google Scholar] [CrossRef]
- Ng, R.K.; Kong, C.T.; So, C.C.; Lui, W.C.; Chan, Y.F.; Leung, K.C.; So, K.C.; Tsang, H.M.; Chan, L.C.; Sham, M.H. Epigenetic dysregulation of leukaemic HOX code in MLL-rearranged leukaemia mouse model. J. Pathol. 2014, 232, 65–74. [Google Scholar] [CrossRef]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2010, 17, 62–68. [Google Scholar] [CrossRef]
- Risner, L.E.; Kuntimaddi, A.; Lokken, A.A.; Achille, N.J.; Birch, N.W.; Schoenfelt, K.; Bushweller, J.H.; Zeleznik-Le, N.J. Functional specificity of CpG DNA-binding CXXC domains in mixed lineage leukemia. J. Biol. Chem. 2013, 288, 29901–29910. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Sato, H.; Wheat, J.C.; Steidl, U.; Ito, K. DNMT3A and TET2 in the pre-leukemic phase of hematopoietic disorders. Front. Oncol. 2016, 6, 187. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Schlenk, R.F.; Paschka, P.; Stolzle, A.; Spath, D.; Kuendgen, A.; von Lilienfeld-Toal, M.; Brugger, W.; Derigs, H.G.; Kremers, S.; et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: Results of the AML Study Group (AMLSG). Blood 2013, 121, 4769–4777. [Google Scholar] [CrossRef]
- Hou, H.A.; Lin, C.C.; Chou, W.C.; Liu, C.Y.; Chen, C.Y.; Tang, J.L.; Lai, Y.J.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia 2014, 28, 50–58. [Google Scholar] [CrossRef]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzen, S.; Hoglund, M.; Bullinger, L.; Dohner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 2017, 168, 801.e13–816.e13. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef]
- Garg, S.; Reyes-Palomares, A.; He, L.; Bergeron, A.; Lavallee, V.P.; Lemieux, S.; Gendron, P.; Rohde, C.; Xia, J.; Jagdhane, P.; et al. Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3-ITD triple-mutated AML. Blood 2019, 134, 263–276. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Tefferi, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Patnaik, M.M.; Hanson, C.A.; Pardanani, A.; Gilliland, D.G.; Levine, R.L. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009, 23, 1343–1345. [Google Scholar] [CrossRef]
- Couronne, L.; Bastard, C.; Bernard, O.A. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Asmar, F.; Punj, V.; Christensen, J.; Pedersen, M.T.; Pedersen, A.; Nielsen, A.B.; Hother, C.; Ralfkiaer, U.; Brown, P.; Ralfkiaer, E.; et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 2013, 98, 1912–1920. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Schnittger, S.; Kohlmann, A.; Kern, W.; Haferlach, T. Mutations of the TET2 and CBL genes: Novel molecular markers in myeloid malignancies. Ann. Hematol. 2010, 89, 643–652. [Google Scholar] [CrossRef]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- National Cancer Research Institute Adult Leukaemia Working Group; Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef]
- Wang, M.L.; Bailey, N.G. Acute myeloid leukemia genetics: Risk stratification and implications for therapy. Arch. Pathol. Lab. Med. 2015, 139, 1215–1223. [Google Scholar] [CrossRef]
- O’Donnell, M.R.; Abboud, C.N.; Altman, J.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; Goorha, S.; et al. NCCN clinical practice guidelines acute myeloid leukemia. J. Natl. Compr. Cancer Netw. 2012, 10, 984–1021. [Google Scholar] [CrossRef]
- Mrozek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in acute leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Akalin, A.; Garrett-Bakelman, F.E.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.A.; Huang, Y.; Biswas, D.; Hess, J.L.; et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar] [CrossRef]
- Bozic, T.; Lin, Q.; Frobel, J.; Wilop, S.; Hoffmann, M.; Muller-Tidow, C.; Brummendorf, T.H.; Jost, E.; Wagner, W. DNA-methylation in C1R is a prognostic biomarker for acute myeloid leukemia. Clin. Epigenet. 2015, 7, 116. [Google Scholar] [CrossRef]
- Lin, T.C.; Hou, H.A.; Chou, W.C.; Ou, D.L.; Yu, S.L.; Tien, H.F.; Lin, L.I. CEBPA methylation as a prognostic biomarker in patients with de novo acute myeloid leukemia. Leukemia 2011, 25, 32–40. [Google Scholar] [CrossRef]
- Jost, E.; Lin, Q.; Weidner, C.I.; Wilop, S.; Hoffmann, M.; Walenda, T.; Schemionek, M.; Herrmann, O.; Zenke, M.; Brummendorf, T.H.; et al. Epimutations mimic genomic mutations of DNMT3A in acute myeloid leukemia. Leukemia 2014, 28, 1227–1234. [Google Scholar] [CrossRef]
- Tao, Y.F.; Fang, F.; Hu, S.Y.; Lu, J.; Cao, L.; Zhao, W.L.; Xiao, P.F.; Li, Z.H.; Wang, N.N.; Xu, L.X.; et al. Hypermethylation of the GATA binding protein 4 (GATA4) promoter in Chinese pediatric acute myeloid leukemia. BMC Cancer 2015, 15, 756. [Google Scholar] [CrossRef]
- Zhou, J.D.; Yao, D.M.; Zhang, Y.Y.; Ma, J.C.; Wen, X.M.; Yang, J.; Guo, H.; Chen, Q.; Lin, J.; Qian, J. GPX3 hypermethylation serves as an independent prognostic biomarker in non-M3 acute myeloid leukemia. Am. J. Cancer Res. 2015, 5, 1786–1794. [Google Scholar]
- Lian, X.Y.; Ma, J.C.; Zhou, J.D.; Zhang, T.J.; Wu, D.H.; Deng, Z.Q.; Zhang, Z.H.; Li, X.X.; He, P.F.; Yan, Y.; et al. Hypermethylation of ITGBL1 is associated with poor prognosis in acute myeloid leukemia. J. Cell. Physiol. 2019, 234, 9438–9446. [Google Scholar] [CrossRef]
- Sellers, Z.P.; Bolkun, L.; Kloczko, J.; Wojtaszewska, M.L.; Lewandowski, K.; Moniuszko, M.; Ratajczak, M.Z.; Schneider, G. Increased methylation upstream of the MEG3 promotor is observed in acute myeloid leukemia patients with better overall survival. Clin. Epigenet. 2019, 11, 50. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, X.; Kajigaya, S.; Cantilena, C.R.; Strickland, S.; Savani, B.N.; Mohan, S.; Feng, X.; Keyvanfar, K.; Dunavin, N.; et al. Epigenetic landscape of the TERT promoter: A potential biomarker for high risk AML/MDS. Br. J. Haematol. 2016, 175, 427–439. [Google Scholar] [CrossRef]
- Deneberg, S.; Guardiola, P.; Lennartsson, A.; Qu, Y.; Gaidzik, V.; Blanchet, O.; Karimi, M.; Bengtzen, S.; Nahi, H.; Uggla, B.; et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood 2011, 118, 5573–5582. [Google Scholar] [CrossRef]
- Marcucci, G.; Yan, P.; Maharry, K.; Frankhouser, D.; Nicolet, D.; Metzeler, K.H.; Kohlschmidt, J.; Mrozek, K.; Wu, Y.Z.; Bucci, D.; et al. Epigenetics meets genetics in acute myeloid leukemia: Clinical impact of a novel seven-gene score. J. Clin. Oncol. 2014, 32, 548–556. [Google Scholar] [CrossRef]
- Kroeger, H.; Jelinek, J.; Estecio, M.R.; He, R.; Kondo, K.; Chung, W.; Zhang, L.; Shen, L.; Kantarjian, H.M.; Bueso-Ramos, C.E.; et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008, 112, 1366–1373. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Xu, Q.; Lv, N.; Jing, Y.; Wang, L.; Wang, X.; Guo, J.; Zhou, L.; Liu, J.; et al. Detection of prognostic methylation markers by methylC-capture sequencing in acute myeloid leukemia. Oncotarget 2017, 8, 110444–110459. [Google Scholar] [CrossRef]
- Jiang, D.; Hong, Q.; Shen, Y.; Xu, Y.; Zhu, H.; Li, Y.; Xu, C.; Ouyang, G.; Duan, S. The diagnostic value of DNA methylation in leukemia: A systematic review and meta-analysis. PLoS ONE 2014, 9, e96822. [Google Scholar] [CrossRef]
- Bullinger, L.; Ehrich, M.; Dohner, K.; Schlenk, R.F.; Dohner, H.; Nelson, M.R.; van den Boom, D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood 2010, 115, 636–642. [Google Scholar] [CrossRef]
- Deneberg, S.; Grovdal, M.; Karimi, M.; Jansson, M.; Nahi, H.; Corbacioglu, A.; Gaidzik, V.; Dohner, K.; Paul, C.; Ekstrom, T.J.; et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia 2010, 24, 932–941. [Google Scholar] [CrossRef]
- Ho, P.A.; Kutny, M.A.; Alonzo, T.A.; Gerbing, R.B.; Joaquin, J.; Raimondi, S.C.; Gamis, A.S.; Meshinchi, S. Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2011, 57, 204–209. [Google Scholar] [CrossRef]
- Farrar, J.E.; Schuback, H.L.; Ries, R.E.; Wai, D.; Hampton, O.A.; Trevino, L.R.; Alonzo, T.A.; Guidry Auvil, J.M.; Davidsen, T.M.; Gesuwan, P.; et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res. 2016, 76, 2197–2205. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Wertheim, G.B.; Smith, C.; Figueroa, M.E.; Kalos, M.; Bagg, A.; Carroll, M.; Master, S.R. Microsphere-based multiplex analysis of DNA methylation in acute myeloid leukemia. J. Mol. Diagn. 2014, 16, 207–215. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Luskin, M.R.; Carroll, M.; Smith, C.; Harrison, J.; Pierce, S.; Kornblau, S.; Konopleva, M.; Kadia, T.; Kantarjian, H.; et al. Validation of a clinical assay of multi-locus DNA methylation for prognosis of newly diagnosed AML. Am. J. Hematol. 2017, 92, E14–E15. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Y.; Zhang, Z.; Li, J.; Wan, Y.; Zhang, L.; Wang, Y.; Li, X.; Xu, Y.; Fu, X.; et al. 5-hydroxymethylcytosine loss is associated with poor prognosis for patients with WHO grade II diffuse astrocytomas. Sci. Rep. 2016, 6, 20882. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, J.; Guo, Z.; Ma, Q.; Xu, Z.; Zhou, Y.; Xu, Z.; Li, Z.; Liu, Y.; Ye, X.; et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016, 26, 103–118. [Google Scholar] [CrossRef]
- Shi, X.; Yu, Y.; Luo, M.; Zhang, Z.; Shi, S.; Feng, X.; Chen, Z.; He, J. Loss of 5-hydroxymethylcytosine is an independent unfavorable prognostic factor for esophageal squamous cell carcinoma. PLoS ONE 2016, 11, e0153100. [Google Scholar] [CrossRef]
- Kroeze, L.I.; Aslanyan, M.G.; van Rooij, A.; Koorenhof-Scheele, T.N.; Massop, M.; Carell, T.; Boezeman, J.B.; Marie, J.P.; Halkes, C.J.; de Witte, T.; et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood 2014, 124, 1110–1118. [Google Scholar] [CrossRef]
- Ahn, J.S.; Kim, H.J.; Kim, Y.K.; Lee, S.S.; Ahn, S.Y.; Jung, S.H.; Yang, D.H.; Lee, J.J.; Park, H.J.; Choi, S.H.; et al. 5-Hydroxymethylcytosine correlates with epigenetic regulatory mutations, but may not have prognostic value in predicting survival in normal karyotype acute myeloid leukemia. Oncotarget 2017, 8, 8305–8314. [Google Scholar] [CrossRef][Green Version]
- Aslanyan, M.G.; Kroeze, L.I.; Langemeijer, S.M.; Koorenhof-Scheele, T.N.; Massop, M.; van Hoogen, P.; Stevens-Linders, E.; van de Locht, L.T.; Tonnissen, E.; van der Heijden, A.; et al. Clinical and biological impact of TET2 mutations and expression in younger adult AML patients treated within the EORTC/GIMEMA AML-12 clinical trial. Ann. Hematol. 2014, 93, 1401–1412. [Google Scholar] [CrossRef]
- Rowe, J.M.; Tallman, M.S. How I treat acute myeloid leukemia. Blood 2010, 116, 3147–3156. [Google Scholar] [CrossRef]
- Yanada, M.; Mori, J.; Aoki, J.; Masuko, M.; Harada, K.; Uchida, N.; Doki, N.; Fukuda, T.; Sakura, T.; Kanamori, H.; et al. Allogeneic hematopoietic cell transplantation for patients with a history of multiple relapses of acute myeloid leukemia. Ann. Hematol. 2019, 98, 2179–2186. [Google Scholar] [CrossRef]
- Deschler, B.; de Witte, T.; Mertelsmann, R.; Lubbert, M. Treatment decision-making for older patients with high-risk myelodysplastic syndrome or acute myeloid leukemia: Problems and approaches. Haematologica 2006, 91, 1513–1522. [Google Scholar]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Lowenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef]
- Sorm, F.; Vesely, J. Effect of 5-aza-2′-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma 1968, 15, 339–343. [Google Scholar]
- Sorm, F.; Piskala, A.; Cihak, A.; Vesely, J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20, 202–203. [Google Scholar] [CrossRef]
- Al-Ali, H.K.; Jaekel, N.; Niederwieser, D. The role of hypomethylating agents in the treatment of elderly patients with AML. J. Geriatr. Oncol. 2014, 5, 89–105. [Google Scholar] [CrossRef]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef]
- Stresemann, C.; Bokelmann, I.; Mahlknecht, U.; Lyko, F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol. Cancer Ther. 2008, 7, 2998–3005. [Google Scholar] [CrossRef]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lubbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef]
- Chang, E.; Ganguly, S.; Rajkhowa, T.; Gocke, C.D.; Levis, M.; Konig, H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia 2016, 30, 1025–1032. [Google Scholar] [CrossRef]
- Lakshmikuttyamma, A.; Scott, S.A.; DeCoteau, J.F.; Geyer, C.R. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene 2010, 29, 576–588. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D.; Abraham, I.; Robertson, K.D.; Fernandez-Zapico, M.E.; Patnaik, M.M. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: A systematic review of hypomethylating agents trials. Clin. Epigenet. 2016, 8, 68. [Google Scholar] [CrossRef]
- Issa, J.P. DNA methylation as a therapeutic target in cancer. Clin. Cancer Res. 2007, 13, 1634–1637. [Google Scholar] [CrossRef]
- Fandy, T.E.; Herman, J.G.; Kerns, P.; Jiemjit, A.; Sugar, E.A.; Choi, S.H.; Yang, A.S.; Aucott, T.; Dauses, T.; Odchimar-Reissig, R.; et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009, 114, 2764–2773. [Google Scholar] [CrossRef]
- Shen, L.; Kantarjian, H.; Guo, Y.; Lin, E.; Shan, J.; Huang, X.; Berry, D.; Ahmed, S.; Zhu, W.; Pierce, S.; et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J. Clin. Oncol. 2010, 28, 605–613. [Google Scholar] [CrossRef]
- Issa, J.P.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef]
- Kamali Dolatabadi, E.; Ostadali Dehaghi, M.; Amirizadeh, N.; Parivar, K.; Mahdian, R. CDKN2B methylation correlates with survival in AML patients. Iran. J. Pharm. Res. 2017, 16, 1600–1611. [Google Scholar]
- Gore, S.D.; Baylin, S.; Sugar, E.; Carraway, H.; Miller, C.B.; Carducci, M.; Grever, M.; Galm, O.; Dauses, T.; Karp, J.E.; et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006, 66, 6361–6369. [Google Scholar] [CrossRef]
- Daskalakis, M.; Nguyen, T.T.; Nguyen, C.; Guldberg, P.; Kohler, G.; Wijermans, P.; Jones, P.A.; Lubbert, M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 2002, 100, 2957–2964. [Google Scholar] [CrossRef]
- Kantarjian, H.; Oki, Y.; Garcia-Manero, G.; Huang, X.; O’Brien, S.; Cortes, J.; Faderl, S.; Bueso-Ramos, C.; Ravandi, F.; Estrov, Z.; et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007, 109, 52–57. [Google Scholar] [CrossRef]
- Yan, P.; Frankhouser, D.; Murphy, M.; Tam, H.H.; Rodriguez, B.; Curfman, J.; Trimarchi, M.; Geyer, S.; Wu, Y.Z.; Whitman, S.P.; et al. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood 2012, 120, 2466–2474. [Google Scholar] [CrossRef]
- Kirschbaum, M.; Gojo, I.; Goldberg, S.L.; Bredeson, C.; Kujawski, L.A.; Yang, A.; Marks, P.; Frankel, P.; Sun, X.; Tosolini, A.; et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br. J. Haematol. 2014, 167, 185–193. [Google Scholar] [CrossRef]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Bitoun, E.; Oliver, P.L.; Davies, K.E. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 2007, 16, 92–106. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.J. Resistance to FLT3 inhibitors in acute myeloid leukemia: Molecular mechanisms and resensitizing strategies. World J. Clin. Oncol. 2018, 9, 90–97. [Google Scholar] [CrossRef]
- Swords, R.; Freeman, C.; Giles, F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia 2012, 26, 2176–2185. [Google Scholar] [CrossRef]
- Al-Jamal, H.A.; Mat Jusoh, S.A.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef]
- Garz, A.K.; Wolf, S.; Grath, S.; Gaidzik, V.; Habringer, S.; Vick, B.; Rudelius, M.; Ziegenhain, C.; Herold, S.; Weickert, M.T.; et al. Azacitidine combined with the selective FLT3 kinase inhibitor crenolanib disrupts stromal protection and inhibits expansion of residual leukemia-initiating cells in FLT3-ITD AML with concurrent epigenetic mutations. Oncotarget 2017, 8, 108738–108759. [Google Scholar] [CrossRef][Green Version]
- Strati, P.; Kantarjian, H.; Ravandi, F.; Nazha, A.; Borthakur, G.; Daver, N.; Kadia, T.; Estrov, Z.; Garcia-Manero, G.; Konopleva, M.; et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am. J. Hematol. 2015, 90, 276–281. [Google Scholar] [CrossRef]
- Wang, E.S. Incorporating FLT3 inhibitors in the frontline treatment of FLT3 mutant acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 154–162. [Google Scholar] [CrossRef]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Huang, Y.H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene(s) | Sample Size | Sample Type | Molecular Method(s) | Clinical Relevance | Reference |
|---|---|---|---|---|---|
| C1R | 194 AML (from TCGA) for analysis; 146 AML for validation | BM | Pyrosequencing | C1R hypermethylation is correlated with better OS | [83] |
| CEBPA | 193 AML | BM | Bisulfite MassARRAY analysis | CEBPA hypermethylation is correlated with better OS | [84] |
| DNMT3A | 194 AML (from TCGA) | BM | Human Methylation 450K BeadChip | DNMT3A hypermethylation (internal promoter) is correlated with lower EFS and OS | [85] |
| GATA4 | 105 AML | BM | MSP, BSP | GATA4 promoter methylation is correlated with MRD and poor OS | [86] |
| GPX3 | 181 de novo AML | BM | Real-time quantitative MSP | GPX3 hypermethylation is correlated with poor OS in non-APL AML | [87] |
| ITGBL1 | 131 AML | BM | Real-time quantitative MSP, BSP | ITGBL1 hypermethylation is correlated with lower CR rate, poor DFS and OS. | [88] |
| MEG3 | 45 AML | PB | Combined bisulfite restriction analysis | MEG3 hypermethylation is correlated with better OS | [89] |
| TERT | 33 AML & 10 AML/MDS | PB/BM | Pyrosequencing | TERT hypermethylation is correlated with poor OS | [90] |
| PcG targets (CDKN2A, CDH1, HIC1 and CDKN2B) | 118 CN-AML | BM | Human Methylation 27 BeadChip, pyrosequencing | Methylation of PcG targets is correlated with better EFS and OS | [91] |
| CD34, RHOC, SCRN1, F2RL1, FAM92A1, MIR155HG, VWA8 | 134 CN-AML for analysis; 355 CN-AML for validation | BM | MethylCap-Seq | Hypermethylation of the selected genes is correlated with better OS | [92] |
| NOR1, CDH13, p15, NPM2, OLIG2, PGR, HIN1, and SLC26A4 | 30 paired AML (diagnosis and relapse) | BM/PB | Pyrosequencing | All the selected genes have increased methylation at relapse | [93] |
| BARD1, BCL9L, CLEC11A, DEFB1, FOXD2, IGF1, IL18, ITIH1, LSP1, P2RX6, RNASE, TUBGCP2 | 21 de novo AML for analysis; 169 from TCGA for validation | BM | MethylCap-Seq | High M-value is correlated with lower CR, increased hazard for DFS, and poor OS | [94] |
| BTBD3, CXCR5, E2F1, FAM110A, FAM30A, GALNT5, KIAA1305, LCK, LMCD1, PRMT7, SLC7A6OS, SMG6, SRR, USP50, VWF, ZFP161 | 344 AML | BM | HELP | Distinct DNA methylation patterns defines new AML subtypes. Methylation of the selected genes is predictive of OS | [18] |
| CCDC85C, CHL1, ELAVL2, FAM115A, FAM196A, GRP146, GPR6, HELZ2, ID4, IL2RA, KCNG3, LOC254559, LOC284801, NPAS2, PCDHAC2, PROB1, SHISA6, SLC18A3, SOCS2, TRIM67, ZFP42 | 138 paired AML (diagnosis and relapse) | BM | ERRBS | Higher number of loci with differential methylation at diagnosis is correlated with shorter time to relapse | [21] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int. J. Mol. Sci. 2019, 20, 4576. https://doi.org/10.3390/ijms20184576
Yang X, Wong MPM, Ng RK. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. International Journal of Molecular Sciences. 2019; 20(18):4576. https://doi.org/10.3390/ijms20184576
Chicago/Turabian StyleYang, Xianwen, Molly Pui Man Wong, and Ray Kit Ng. 2019. "Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications" International Journal of Molecular Sciences 20, no. 18: 4576. https://doi.org/10.3390/ijms20184576
APA StyleYang, X., Wong, M. P. M., & Ng, R. K. (2019). Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. International Journal of Molecular Sciences, 20(18), 4576. https://doi.org/10.3390/ijms20184576