Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of Niacin on Parkinson’s Disease Subjects and an MPP+-Induced Parkinson’s Disease Model of Neuronal Cell Line N27
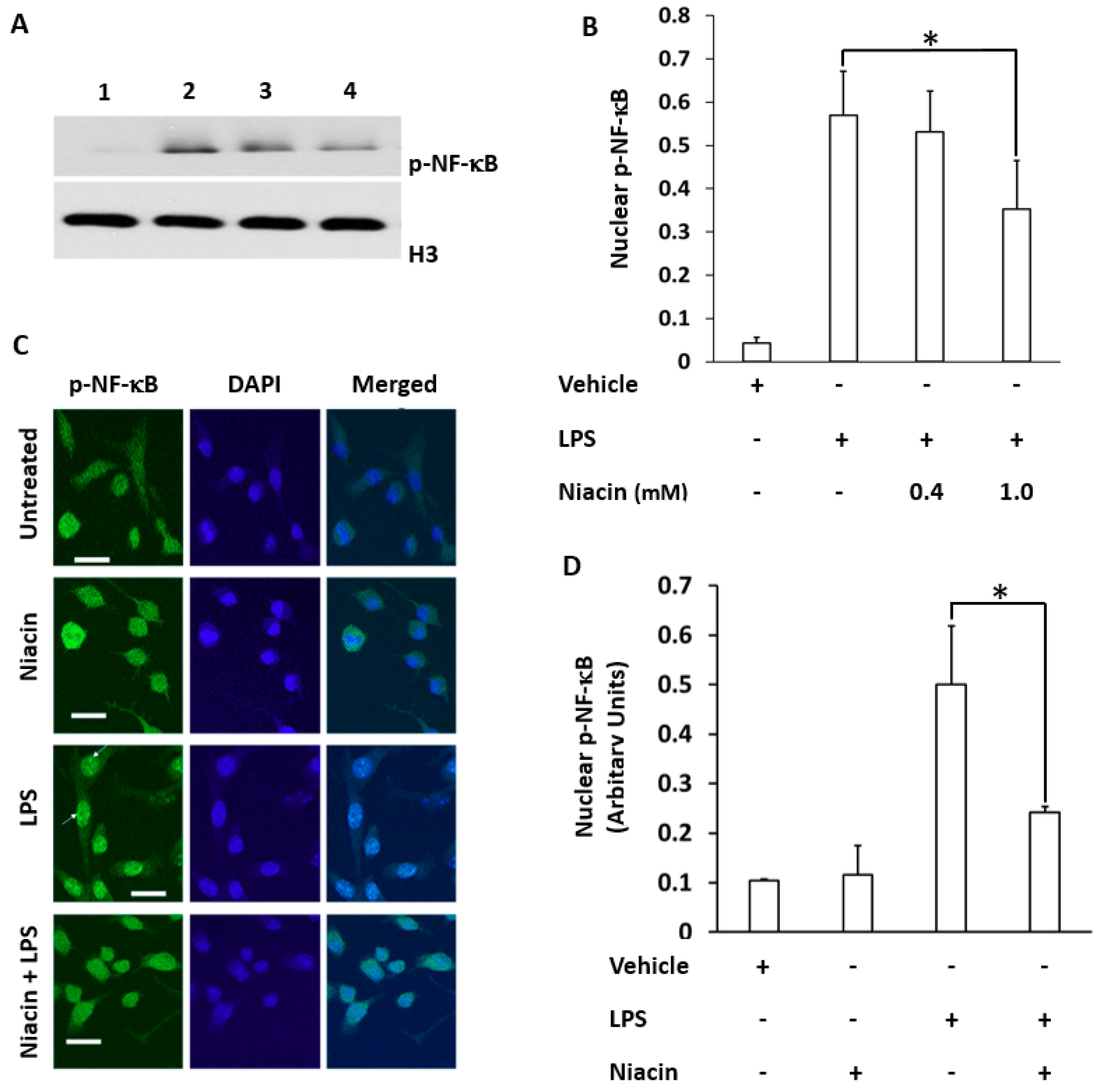
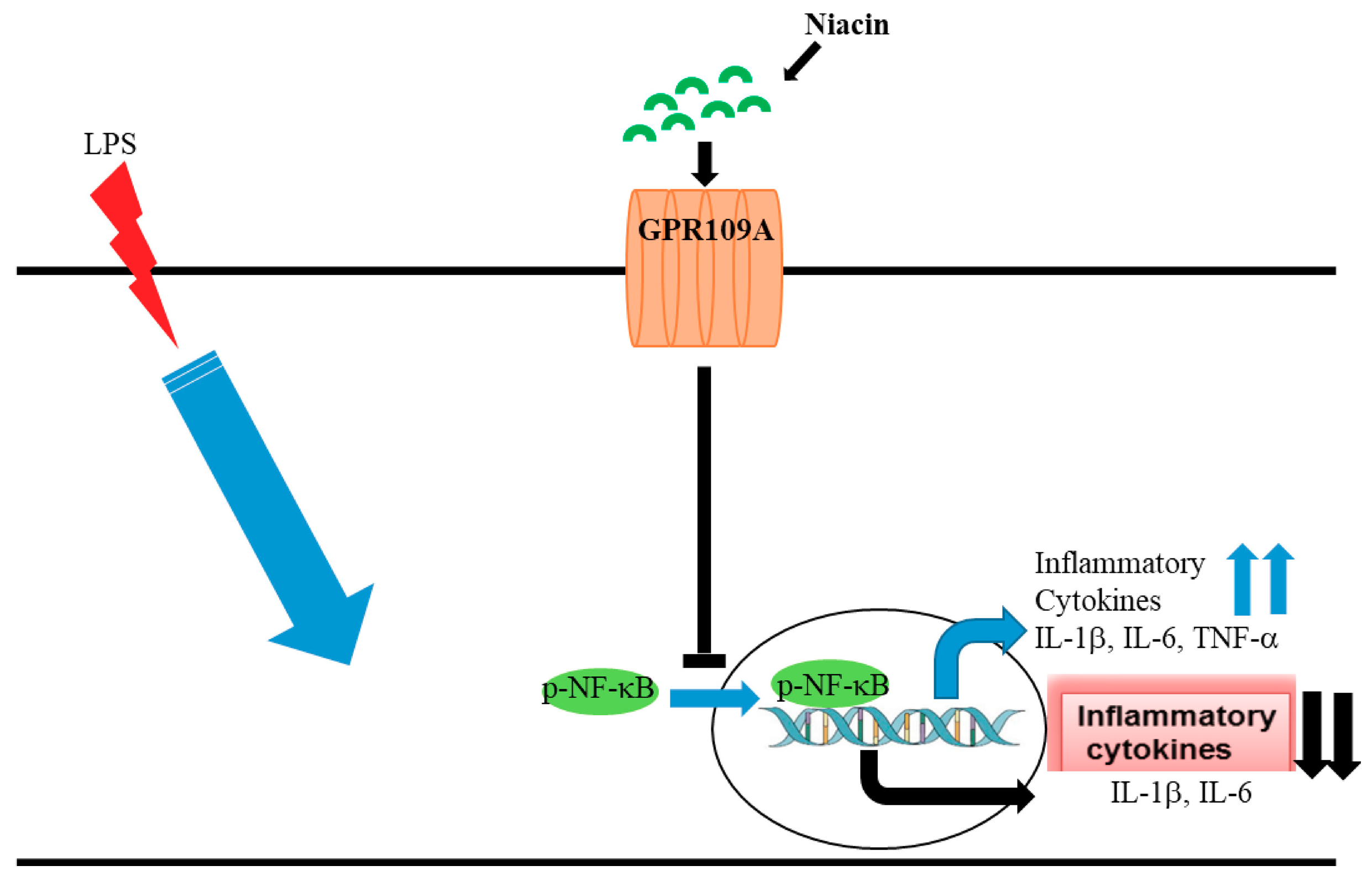
2.2. LPS Induced the Translocation of Phosphorylated Nuclear Factor-κB (p-NF-κB) to the Nucleus Inhibited by Niacin in Cultured RAW264.7 Cells
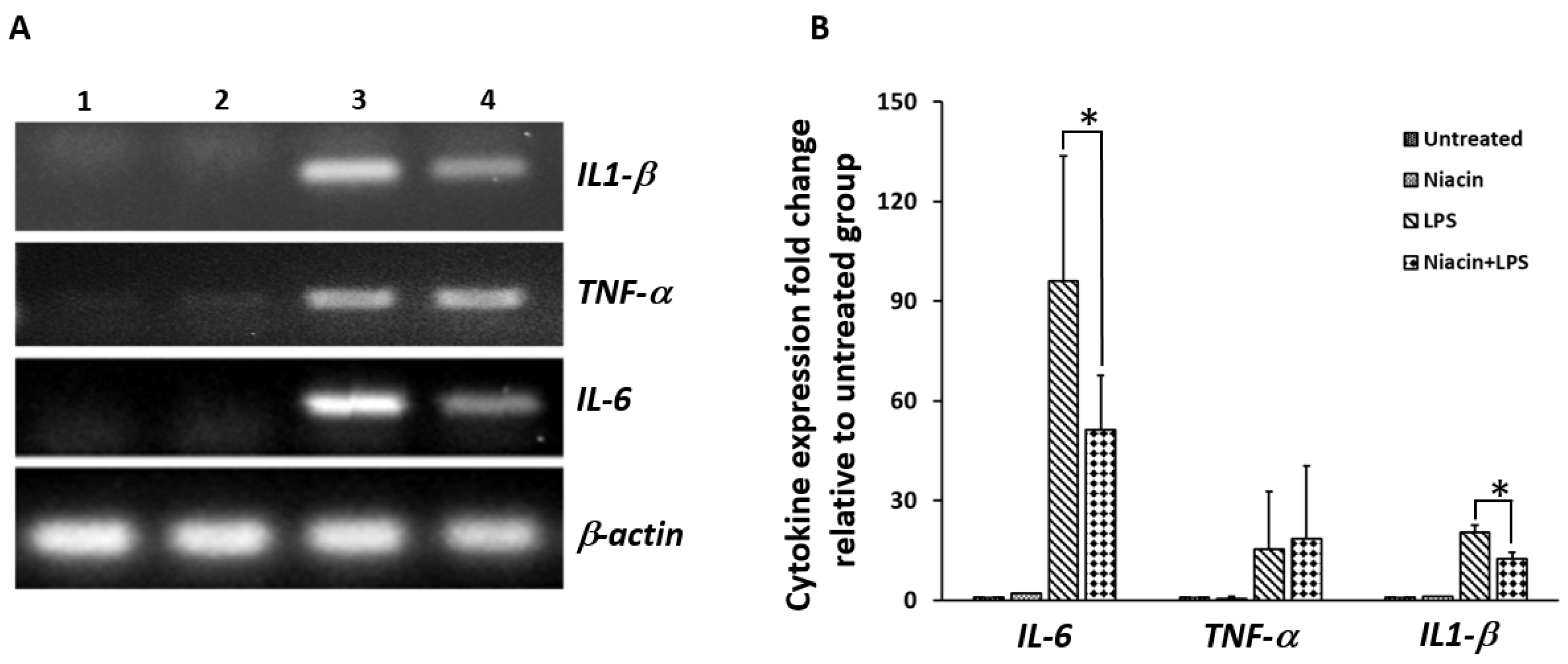
2.3. Effect of Niacin on the Up-Regulation of IL-1β, IL-6, and TNF-α mRNA Induced by LPS
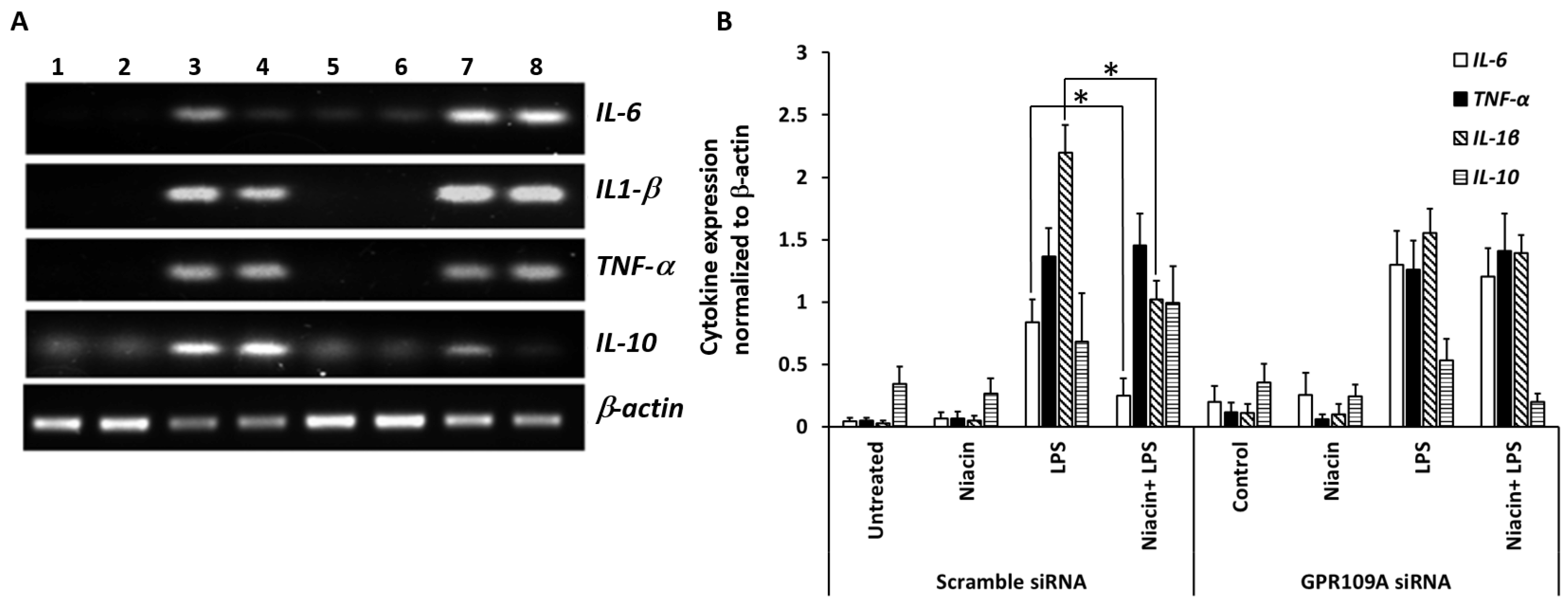
2.4. Silencing of GPR109A by Gene-Specific siRNA in RAW264.7 Cells
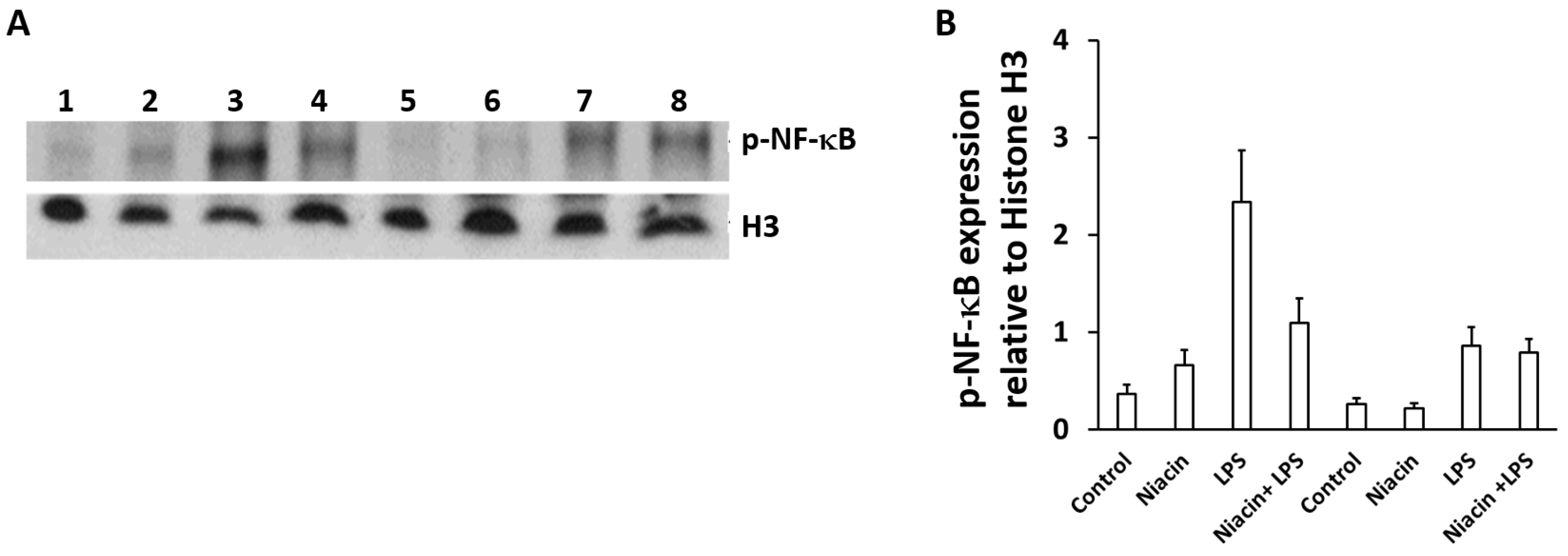
2.5. Niacin Inhibits LPS-Induced Shuttling of p-NF-κB via GPR109A
2.6. Niacin Failed to Inhibit LPS-Induced Inflammation Response in GPR109A Knockdown Cells as Determined by Inflammatory Cytokines
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. PD Subjects
4.3. HPLC Analyses
4.4. Cell Culture
4.5. siRNA Transfection
4.6. Nuclear and Cytosolic Fractions
4.7. Western Blot
4.8. Reverse Transcriptase Polymerase Chain Reaction Analysis
4.9. Immunostaining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPR109A | G-protein-coupled receptor 109A |
| IL-6 | Interleukin 6 |
| LPS | Lipopolysaccharide |
| IL-1β | Interleukin 1β |
| NF-κB | Nuclear factor κB |
| TNF-α | Tumor necrosis factor alpha |
| PD | Parkinson’s disease |
References
- Feingold, K.R.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. Inflammation stimulates niacin receptor (GPR109A/HCA2) expression in adipose tissue and macrophages. J. lipid Res. 2014, 55, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T. Niacin metabolism and Parkinson’s disease. Environ. Health Prev. Med. 2005, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F. Caffeine and Parkinson’s Disease: From Molecular Targets to Epidemiology and Clinical Trials in Coffee: Consumption and Health Implications; Farah, A., Ed.; Royal Society of Chemistry: Croydon, UK, 2019; pp. 171–195. [Google Scholar]
- Wise, A.; Ford, S.M.; Fraser, N.J.; Barnes, A.A.; Elshourbagy, N.; Eilert, M.; Ignar, D.M.; Murdock, P.R.; Steplewski, K.; Green, A.; et al. Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 2003, 278, 9869–9874. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Heng, L.; Shaodong, W.; Fangyu, W.; Zhenkai, W. Pellagra affecting a patient with Crohn’s disease. An. Bras Dermatol. 2017, 92, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kong, D.; Wang, Q.; Wu, W.; Tang, Y.; Bai, T.; Guo, L.; Wei, L.; Zhang, Q.; Yu, Y.; et al. Niacin ameliorates ulcerative colitis via prostaglandin D2-mediated D prostanoid receptor 1 activation. EMBO Mol. Med. 2017, 9, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan Res. 2018, 11, 1178646918776658. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Giri, B.; Malik, A.; Khodadadi, H.; Morgan, J.C.; Chong, R.K.; Baban, B. Niacin modulates macrophage polarization in Parkinson’s disease. J. Neuroimmunol. 2018, 320, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflam. 2017, 2017. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef]
- Maciejewski-Lenoir, D.; Richman, J.G.; Hakak, Y.; Gaidarov, I.; Behan, D.P.; Connolly, D.T. Langerhans cells release prostaglandin D2 in response to nicotinic acid. J. Investig. Dermatol. 2006, 126, 2637–2646. [Google Scholar] [CrossRef]
- Offermanns, S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharm. Sci. 2006, 27, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S.; Schwaninger, M. Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol. Med. 2015, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Ananth, S.; Cresci, G.; Roon, P.; Smith, S.; Ganapathy, V. Expression and localization of GPR109A (PUMA-G/HM74A) mRNA and protein in mammalian retinal pigment epithelium. Mol. Vis. 2009, 15, 362–372. [Google Scholar] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Itano, Y.; Kubo, T.; Nomura, Y. Suppressive effect of FK-506, a novel immunosuppressant, against MPTP-induced dopamine depletion in the striatum of young C57BL/6 mice. J. Neuroimmunol. 1994, 50, 221–224. [Google Scholar] [CrossRef]
- Kurkowska-Jastrzebska, I.; Wronska, A.; Kohutnicka, M.; Czlonkowski, A.; Czlonkowska, A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp. Neurol. 1999, 156, 50–61. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Serapide, M.F.; Caniglia, S.; Testa, N.; Leggio, L.; Vivarelli, S.; Iraci, N.; Pluchino, S.; Marchetti, B. Microglia Polarization, Gene-Environment Interactions and Wnt/beta-Catenin Signaling: Emerging Roles of Glia-Neuron and Glia-Stem/Neuroprogenitor Crosstalk for Dopaminergic Neurorestoration in Aged Parkinsonian Brain. Front. Aging Neurosci. 2018, 10, 12. [Google Scholar] [CrossRef]
- Francois, A.; Terro, F.; Janet, T.; Rioux Bilan, A.; Paccalin, M.; Page, G. Involvement of interleukin-1beta in the autophagic process of microglia: Relevance to Alzheimer’s disease. J. Neuroinflamm. 2013, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Juskewitch, J.E.; Platt, J.L.; Knudsen, B.E.; Knutson, K.L.; Brunn, G.J.; Grande, J.P. Disparate roles of marrow- and parenchymal cell-derived TLR4 signaling in murine LPS-induced systemic inflammation. Sci. Rep. 2012, 2, 918. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.F.; Sa-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1beta generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–120. [Google Scholar] [CrossRef] [PubMed]
- Bromfield, J.J.; Sheldon, I.M. Lipopolysaccharide initiates inflammation in bovine granulosa cells via the TLR4 pathway and perturbs oocyte meiotic progression in vitro. Endocrinology 2011, 152, 5029–5040. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Chong, R.; Bradley, E.; Morgan, J.C. Low-dose niacin supplementation modulates GPR109A, niacin index and ameliorates Parkinson’s disease symptoms without side effects. Clin. Case Rep. 2015, 3, 635–637. [Google Scholar] [CrossRef]
- Sakai, J.; Cammarota, E.; Wright, J.A.; Cicuta, P.; Gottschalk, R.A.; Li, N.; Fraser, I.D.C.; Bryant, C.E. Lipopolysaccharide-induced NF-kappaB nuclear translocation is primarily dependent on MyD88, but TNFalpha expression requires TRIF and MyD88. Sci. Rep. 2017, 7, 1428. [Google Scholar] [CrossRef]
- More, S.V.; Choi, D.K. Emerging preclinical pharmacological targets for Parkinson’s disease. Oncotarget 2016, 77, 29835–29863. [Google Scholar]
- Lofrumento, D.D.; Saponaro, C.; Cianciulli, A.; De Nuccio, F.; Mitolo, V.; Nicolardi, G.; Panaro, M.A. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 2011, 18, 79–88. [Google Scholar] [CrossRef]
- Ojha, S.; Javed, H.; Azimullah, S.; Abul Khair, S.B.; Haque, M.E. Glycyrrhizic acid Attenuates Neuroinflammation and Oxidative Stress in Rotenone Model of Parkinson’s Disease. Neurotox. Res. 2016, 29, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Digby, J.E.; Martinez, F.; Jefferson, A.; Ruparelia, N.; Chai, J.; Wamil, M.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Sasaki, A.; Yoshimura, A. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 2000, 18, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Shmarina, G.V.; Pukhalsky, A.L.; Kokarovtseva, S.N.; Pukhalskaya, D.A.; Shabalova, L.A.; Kapranov, N.I.; Kashirskaja, N.J. Tumor necrosis factor-alpha/interleukin-10 balance in normal and cystic fibrosis children. Mediat. Inflamm. 2001, 10, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wanidworanun, C.; Strober, W. Predominant role of tumor necrosis factor-alpha in human monocyte IL-10 synthesis. J. Immunol. 1993, 151, 6853–6861. [Google Scholar]
- Digby, J.E.; McNeill, E.; Dyar, O.J.; Lam, V.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in adipocytes demonstrated by suppression of fractalkine, RANTES, and MCP-1 and upregulation of adiponectin. Atherosclerosis 2010, 209, 89–95. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirol. 2002, 8, 529–538. [Google Scholar] [CrossRef]
- Fall, P.A.; Fredrikson, M.; Axelson, O.; Granerus, A.K. Nutritional and occupational factors influencing the risk of Parkinson’s disease: A case-control study in southeastern Sweden. Mov. Disord. 1999, 14, 28–37. [Google Scholar] [CrossRef]
- Antalis, T.M.; Godbolt, D. Isolation of intact nuclei from hematopoietic cell types. Nucleic Acids Res. 1991, 19, 4301. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giri, B.; Belanger, K.; Seamon, M.; Bradley, E.; Purohit, S.; Chong, R.; Morgan, J.C.; Baban, B.; Wakade, C. Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A. Int. J. Mol. Sci. 2019, 20, 4559. https://doi.org/10.3390/ijms20184559
Giri B, Belanger K, Seamon M, Bradley E, Purohit S, Chong R, Morgan JC, Baban B, Wakade C. Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A. International Journal of Molecular Sciences. 2019; 20(18):4559. https://doi.org/10.3390/ijms20184559
Chicago/Turabian StyleGiri, Banabihari, Kasey Belanger, Marissa Seamon, Eric Bradley, Sharad Purohit, Raymond Chong, John C. Morgan, Babak Baban, and Chandramohan Wakade. 2019. "Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A" International Journal of Molecular Sciences 20, no. 18: 4559. https://doi.org/10.3390/ijms20184559
APA StyleGiri, B., Belanger, K., Seamon, M., Bradley, E., Purohit, S., Chong, R., Morgan, J. C., Baban, B., & Wakade, C. (2019). Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A. International Journal of Molecular Sciences, 20(18), 4559. https://doi.org/10.3390/ijms20184559