Heat Shock Proteins and Inflammasomes


Abstract
1. Introduction
1.1. HSP
1.1.1. HSP100
1.1.2. HSP90
1.1.3. HSP70
1.1.4. HSP60
1.1.5. HSP40
1.1.6. sHSP
1.2. Inflammasomes
1.2.1. NLRA
1.2.2. NLRB
1.2.3. NLRC
1.2.4. NLRP
2. Positive Effects of HSP on Inflammasomes Activation
2.1. HSP60
2.2. HSP70
2.3. HSP90
2.4. Gp96
3. Negative Effects of HSP on Inflammasomes Activation
3.1. HSP27
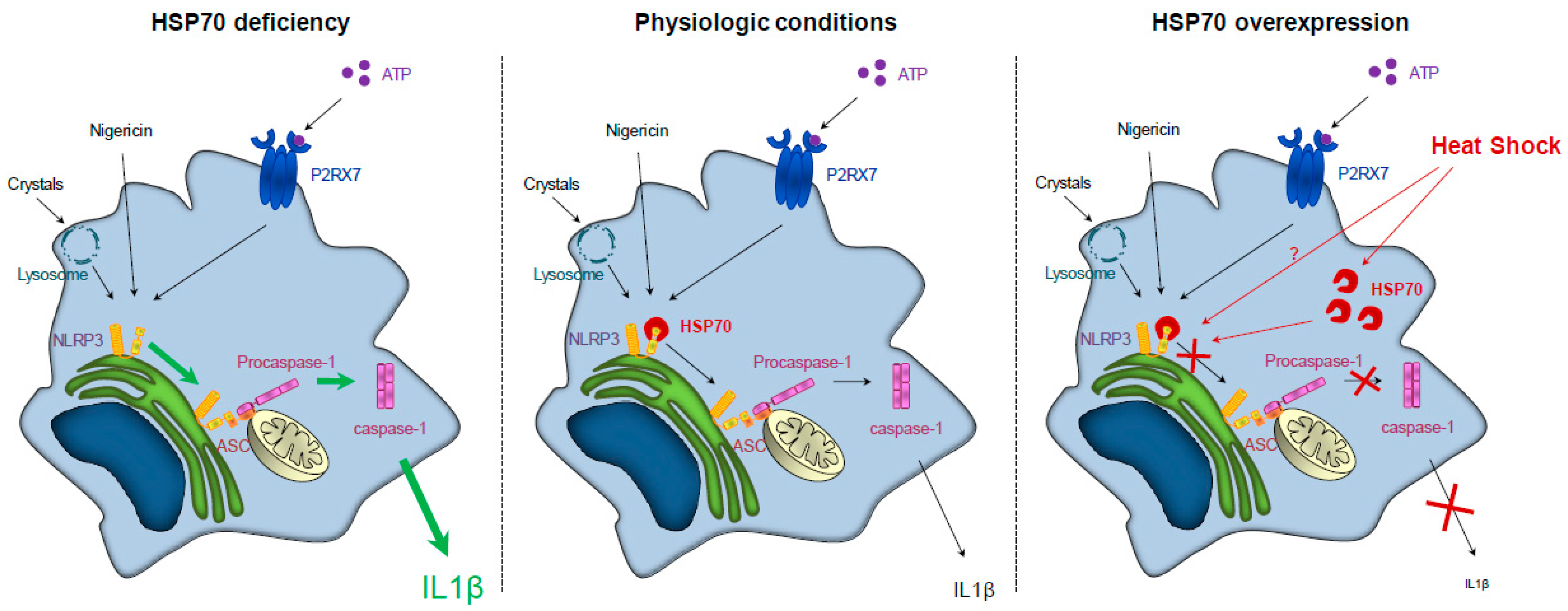
3.2. HSP70
4. Impact of HSP Polymorphisms on Inflammatory Diseases Development
5. Modulation of HSP Expression or Activity in Inflammatory Diseases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mogk, A.; Kummer, E.; Bukau, B. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front. Mol. Biosci. 2015, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, B.; Dai, J.; Srivastava, P.K.; Zammit, D.J.; Lefrancois, L.; Li, Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007, 26, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C. Mechanisms of the Hsp70 chaperone system. Biochem. Cell Biol. 2010, 88, 291–300. [Google Scholar] [CrossRef]
- Sharma, S.K.; De los Rios, P.; Christen, P.; Lustig, A.; Goloubinoff, P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 2010, 6, 914–920. [Google Scholar] [CrossRef]
- Rothnie, A.; Clarke, A.R.; Kuzmic, P.; Cameron, A.; Smith, C.J. A sequential mechanism for clathrin cage disassembly by 70-kDa heat-shock cognate protein (Hsc70) and auxilin. Proc. Natl. Acad. Sci. USA 2011, 108, 6927–6932. [Google Scholar] [CrossRef]
- Mattoo, R.U.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamamoto, H.; Kudo, I.; Matsumoto, K.; Odaka, M.; Grave, E.; Itoh, H. HSP60 possesses a GTPase activity and mediates protein folding with HSP10. Sci. Rep. 2017, 7, 16931. [Google Scholar] [CrossRef]
- Bascos, N.A.D.; Mayer, M.P.; Bukau, B.; Landry, S.J. The Hsp40 J-domain modulates Hsp70 conformation and ATPase activity with a semi-elliptical spring. Protein. Sci. 2017, 26, 1838–1851. [Google Scholar] [CrossRef]
- Kriehuber, T.; Rattei, T.; Weinmaier, T.; Bepperling, A.; Haslbeck, M.; Buchner, J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010, 24, 3633–3642. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J. Alpha-crystallin. Exp. Eye Res. 2003, 76, 145–153. [Google Scholar] [CrossRef]
- Van Montfort, R.; Slingsby, C.; Vierling, E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein. Chem. 2001, 59, 105–156. [Google Scholar] [PubMed]
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Schlieker, C.; Friedrich, K.L.; Schonfeld, H.J.; Vierling, E.; Bukau, B. Refolding of substrates bound to small HSP relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J. Biol. Chem. 2003, 278, 31033–31042. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Miess, A.; Stromer, T.; Walter, S.; Buchner, J. Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J. Biol. Chem. 2005, 280, 23861–23868. [Google Scholar] [CrossRef] [PubMed]
- Cashikar, A.G.; Duennwald, M.; Lindquist, S.L. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J. Biol. Chem. 2005, 280, 23869–23875. [Google Scholar] [CrossRef] [PubMed]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Cycon, K.A.; Mulvaney, K.; Rimsza, L.M.; Persky, D.; Murphy, S.P. Histone deacetylase inhibitors activate CIITA and MHC class II antigen expression in diffuse large B-cell lymphoma. Immunology 2013, 140, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Halff, E.F.; Diebolder, C.A.; Versteeg, M.; Schouten, A.; Brondijk, T.H.; Huizinga, E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 2012, 287, 38460–38472. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011, 477, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci. Rep. 2012, 32, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, T.; Lian, D.; McCully, M.L.; Garcia, B.; Zhong, R.; Madrenas, J. RIP2 is required for NOD signaling but not for Th1 cell differentiation and cellular allograft rejection. Am. J. Transplant. 2008, 8, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Zimmermann, A.G.; Roberts, R.A.; Zhang, L.; Swanson, K.V.; Wen, H.; Davis, B.K.; Allen, I.C.; Holl, E.K.; Ye, Z.; et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat. Immunol. 2012, 13, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Cheng, G. NOD so fast: NLRX1 puts the brake on inflammation. Immunity 2011, 34, 821–822. [Google Scholar] [CrossRef][Green Version]
- Kersse, K.; Bertrand, M.J.; Lamkanfi, M.; Vandenabeele, P. NOD-like receptors and the innate immune system: Coping with danger, damage and death. Cytokine Growth Factor Rev. 2011, 22, 257–276. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Brunner, R.M.; Goldammer, T.; Kuhn, C.; Walz, C.; Chomdej, S.; Tesfaye, D.; Schellander, K.; Wimmers, K.; Schwerin, M. Bovine NALP5, NALP8, and NALP9 genes: Assignment to a QTL region and the expression in adult tissues, oocytes, and preimplantation embryos. Biol. Reprod. 2006, 74, 577–584. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Westerveld, G.H.; Korver, C.M.; van Pelt, A.M.; Leschot, N.J.; van der Veen, F.; Repping, S.; Lombardi, M.P. Mutations in the testis-specific NALP14 gene in men suffering from spermatogenic failure. Hum. Reprod. 2006, 21, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Pascal, G.; Monget, P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol. Biol. 2009, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Imamura, R.; Wang, Y.; Kinoshita, T.; Suzuki, M.; Noda, T.; Sagara, J.; Taniguchi, S.; Okamoto, H.; Suda, T. Anti-inflammatory activity of PYNOD and its mechanism in humans and mice. J. Immunol. 2010, 184, 5874–5884. [Google Scholar] [CrossRef]
- Joly, S.; Eisenbarth, S.C.; Olivier, A.K.; Williams, A.; Kaplan, D.H.; Cassel, S.L.; Flavell, R.A.; Sutterwala, F.S. Cutting edge: Nlrp10 is essential for protective antifungal adaptive immunity against Candida albicans. J. Immunol. 2012, 189, 4713–4717. [Google Scholar] [CrossRef] [PubMed]
- Damm, A.; Lautz, K.; Kufer, T.A. Roles of NLRP10 in innate and adaptive immunity. Microbes. Infect. 2013, 15, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Peng, H.; Chang, B.; Lu, C.; Su, J.; Wu, Y.; Lv, P.; Wang, Y.; Liu, J.; Zhang, B.; Quan, F.; et al. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS ONE 2012, 7, e30344. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; Grandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Lich, J.D.; Ye, Z.; Allen, I.C.; Gris, D.; Wilson, J.E.; Schneider, M.; Roney, K.E.; O’Connor, B.P.; Moore, C.B.; et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J. Immunol. 2010, 185, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Adlakha, Y.K.; Basu, A. HSP60 critically regulates endogenous IL-1beta production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflamm. 2018, 15, 177. [Google Scholar] [CrossRef]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Khalafalla, M.G.; Woods, L.T.; Camden, J.M.; Khan, A.A.; Limesand, K.H.; Petris, M.J.; Erb, L.; Weisman, G.A. P2X7 receptor antagonism prevents IL-1beta release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. J. Biol. Chem. 2017, 292, 16626–16637. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [PubMed]
- Levin, T.C.; Wickliffe, K.E.; Leppla, S.H.; Moayeri, M. Heat shock inhibits caspase-1 activity while also preventing its inflammasome-mediated activation by anthrax lethal toxin. Cell. Microbiol. 2008, 10, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Wang, J.; Liao, F.; Yan, X.; Li, J.; Huang, L.; Liu, F. Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2X7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis After Subarachnoid Hemorrhage in Mice. Front. Mol. Neurosci. 2018, 11, 401. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, X.; Su, G.; Wang, Y.; Wang, Z.; Qing, S.; Jia, J.; Wang, Y.; Huang, L.; Zheng, K.; et al. AT-533, a Hsp90 inhibitor, attenuates HSV-1-induced inflammation. Biochem. Pharmacol. 2019, 166, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sedlacek, A.L.; Pawaria, S.; Xu, H.; Scott, M.J.; Binder, R.J. Cutting Edge: The Heat Shock Protein gp96 Activates Inflammasome-Signaling Platforms in APCs. J. Immunol. 2018, 201, 2209–2214. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.; Chen, Y.X.; McNulty, M.; Simard, T.; Zhao, X.; Wells, D.J.; de Belleroche, J.; O’Brien, E.R. Extracellular release of the atheroprotective heat shock protein 27 is mediated by estrogen and competitively inhibits acLDL binding to scavenger receptor-A. Circ. Res. 2008, 103, 133–141. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Dodd, S.L.; Hain, B.; Senf, S.M.; Judge, A.R. Hsp27 inhibits IKKbeta-induced NF-kappaB activity and skeletal muscle atrophy. FASEB J. 2009, 23, 3415–3423. [Google Scholar] [CrossRef]
- You, W.; Min, X.; Zhang, X.; Qian, B.; Pang, S.; Ding, Z.; Li, C.; Gao, X.; Di, R.; Cheng, Y.; et al. Cardiac-specific expression of heat shock protein 27 attenuated endotoxin-induced cardiac dysfunction and mortality in mice through a PI3K/Akt-dependent mechanism. Shock 2009, 32, 108–117. [Google Scholar] [CrossRef]
- Chen, Y.; Ross, B.M.; Currie, R.W. Heat shock treatment protects against angiotensin II-induced hypertension and inflammation in aorta. Cell Stress Chaperones 2004, 9, 99–107. [Google Scholar] [CrossRef]
- Hadadi, E.; Zhang, B.; Baidzajevas, K.; Yusof, N.; Puan, K.J.; Ong, S.M.; Yeap, W.H.; Rotzschke, O.; Kiss-Toth, E.; Wilson, H.; et al. Differential IL-1beta secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci. Rep. 2016, 6, 39035. [Google Scholar] [CrossRef]
- Martine, P.; Chevriaux, A.; Derangere, V.; Apetoh, L.; Garrido, C.; Ghiringhelli, F.; Rebe, C. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Chuang, E.; Hori, A.M.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, jcs189928. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 10480–10485. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Petrilli, V. The multifaceted roles of inflammasome proteins in cancer. Curr. Opin. Oncol. 2017, 29, 35–40. [Google Scholar] [CrossRef]
- Aganna, E.; Martinon, F.; Hawkins, P.N.; Ross, J.B.; Swan, D.C.; Booth, D.R.; Lachmann, H.J.; Bybee, A.; Gaudet, R.; Woo, P.; et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002, 46, 2445–2452. [Google Scholar] [CrossRef]
- Boiocchi, C.; Osera, C.; Monti, M.C.; Ferraro, O.E.; Govoni, S.; Cuccia, M.; Montomoli, C.; Pascale, A.; Bergamaschi, R. Are Hsp70 protein expression and genetic polymorphism implicated in multiple sclerosis inflammation? J. Neuroimmunol. 2014, 268, 84–88. [Google Scholar] [CrossRef]
- Klausz, G.; Molnar, T.; Nagy, F.; Gyulai, Z.; Boda, K.; Lonovics, J.; Mandi, Y. Polymorphism of the heat-shock protein gene Hsp70-2, but not polymorphisms of the IL-10 and CD14 genes, is associated with the outcome of Crohn’s disease. Scand. J. Gastroenterol. 2005, 40, 1197–1204. [Google Scholar] [CrossRef]
- Debler, J.; Schiemann, U.; Seybold, U.; Mussack, T.; Landauer, N.; Ladurner, R.; Gross, M. Heat-shock protein HSP70-2 genotypes in patients with Crohn’s disease: A more severe clinical course with intestinal complications in presence of the PstI-polymorphism. Eur. J. Med. Res. 2003, 8, 120–124. [Google Scholar]
- Balog, A.; Gyulai, Z.; Boros, L.G.; Farkas, G.; Takacs, T.; Lonovics, J.; Mandi, Y. Polymorphism of the TNF-alpha, HSP70-2, and CD14 genes increases susceptibility to severe acute pancreatitis. Pancreas 2005, 30, e46–e50. [Google Scholar] [CrossRef]
- Furnrohr, B.G.; Wach, S.; Kelly, J.A.; Haslbeck, M.; Weber, C.K.; Stach, C.M.; Hueber, A.J.; Graef, D.; Spriewald, B.M.; Manger, K.; et al. Polymorphisms in the Hsp70 gene locus are genetically associated with systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 1983–1989. [Google Scholar] [CrossRef]
- Huang, M.N.; Yu, H.; Moudgil, K.D. The involvement of heat-shock proteins in the pathogenesis of autoimmune arthritis: A critical appraisal. Semin. Arthritis Rheum. 2010, 40, 164–175. [Google Scholar] [CrossRef]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef]
- Kacimi, R.; Yenari, M.A. Pharmacologic heat shock protein 70 induction confers cytoprotection against inflammation in gliovascular cells. Glia 2015, 63, 1200–1212. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Mjahed, H.; Girodon, F.; Fontenay, M.; Garrido, C. Heat shock proteins in hematopoietic malignancies. Exp. Cell Res. 2012, 318, 1946–1958. [Google Scholar] [CrossRef]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef]


{kind=link}
| HSP | Inflammasome Activation | Inflammasome Inhibition | Mechanism |
|---|---|---|---|
| Ext HSP27 | + | Competition with acLDL | |
| Int HSP27 | + | Unknown | |
| HSP60 | + | NF-κB activation/ROS | |
| Intra HSP70 | + | Interaction with NLRP3 | |
| Ext HSP70 | + | NF-κB activation | |
| HSP90 | + | Stabilization of the complex with SGT1 and interaction with NLRP3 | |
| Gp96 | + | NF-κB activation or K+ efflux |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martine, P.; Rébé, C. Heat Shock Proteins and Inflammasomes. Int. J. Mol. Sci. 2019, 20, 4508. https://doi.org/10.3390/ijms20184508
Martine P, Rébé C. Heat Shock Proteins and Inflammasomes. International Journal of Molecular Sciences. 2019; 20(18):4508. https://doi.org/10.3390/ijms20184508
Chicago/Turabian StyleMartine, Pierre, and Cédric Rébé. 2019. "Heat Shock Proteins and Inflammasomes" International Journal of Molecular Sciences 20, no. 18: 4508. https://doi.org/10.3390/ijms20184508
APA StyleMartine, P., & Rébé, C. (2019). Heat Shock Proteins and Inflammasomes. International Journal of Molecular Sciences, 20(18), 4508. https://doi.org/10.3390/ijms20184508