Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors



 , and
, and 
Abstract
1. Introduction
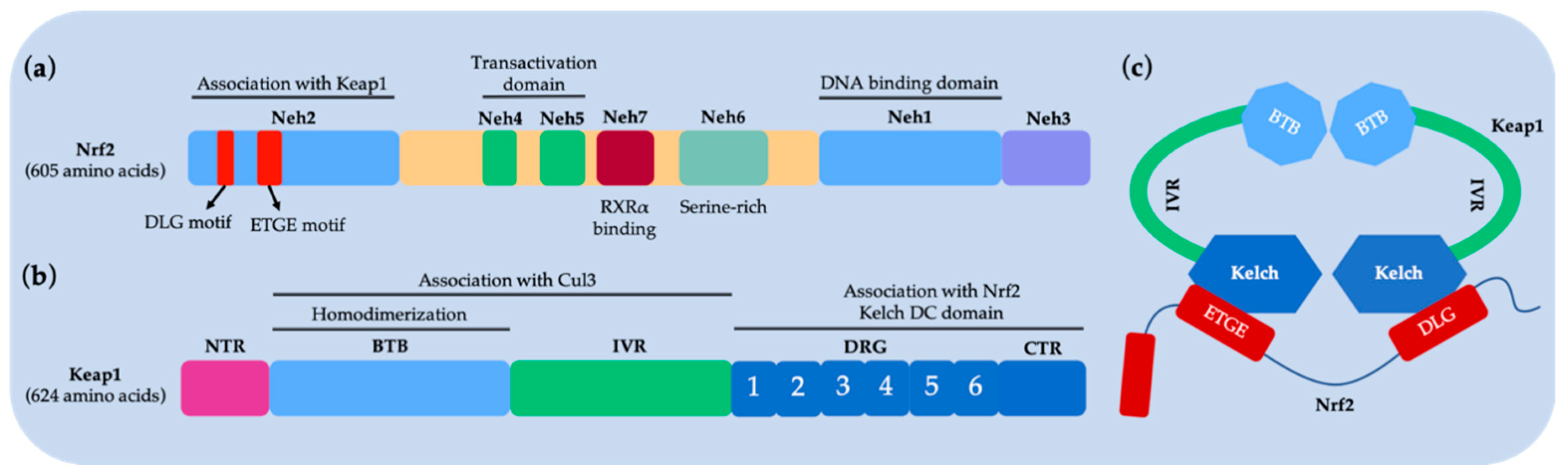
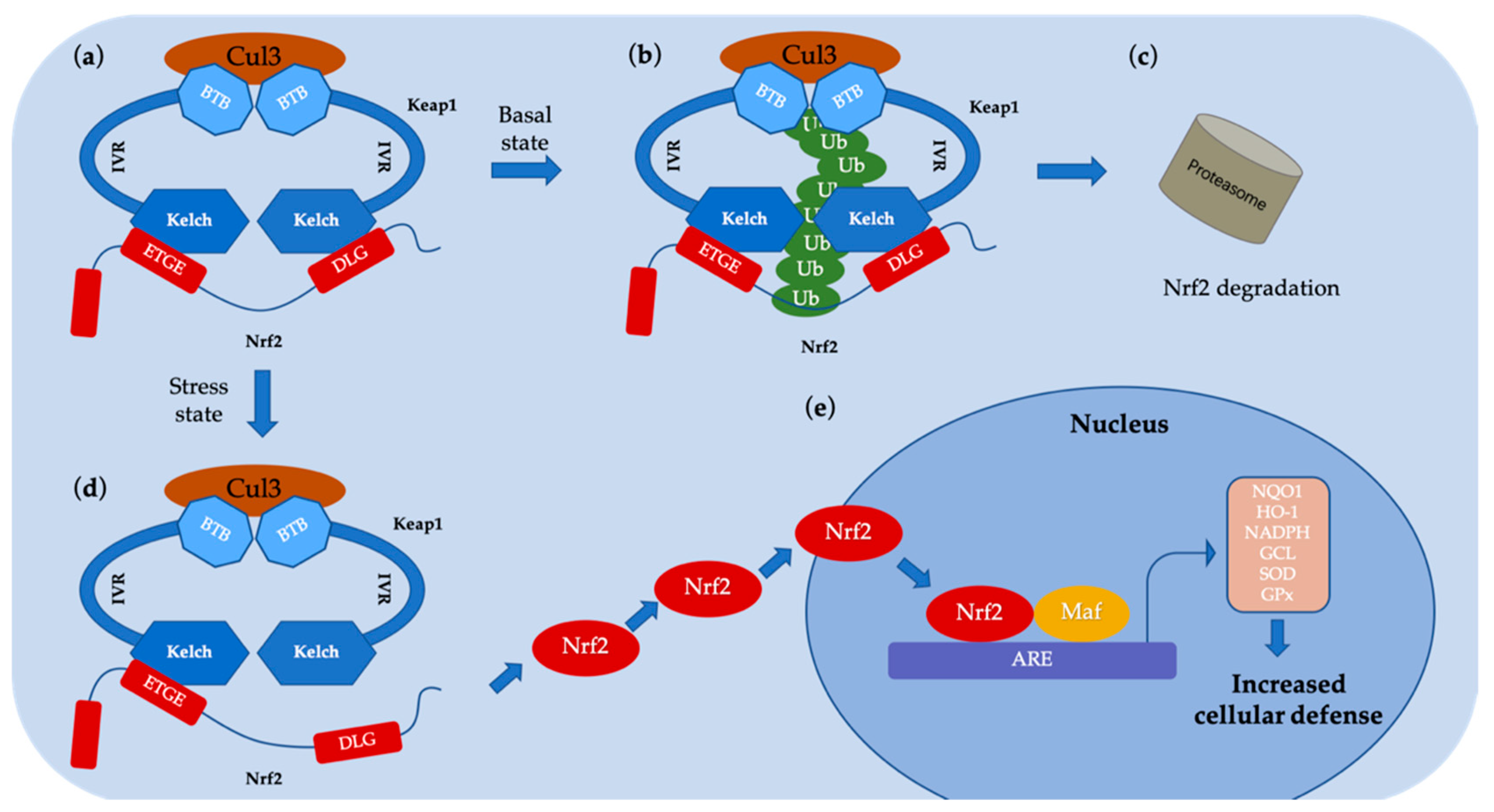
2. Diseases Related to the Nrf2–Keap1 Protein–Protein Interaction
2.1. Cancer
2.2. Neurodegenerative Disease
2.3. Diabetes
2.4. Other Diseases
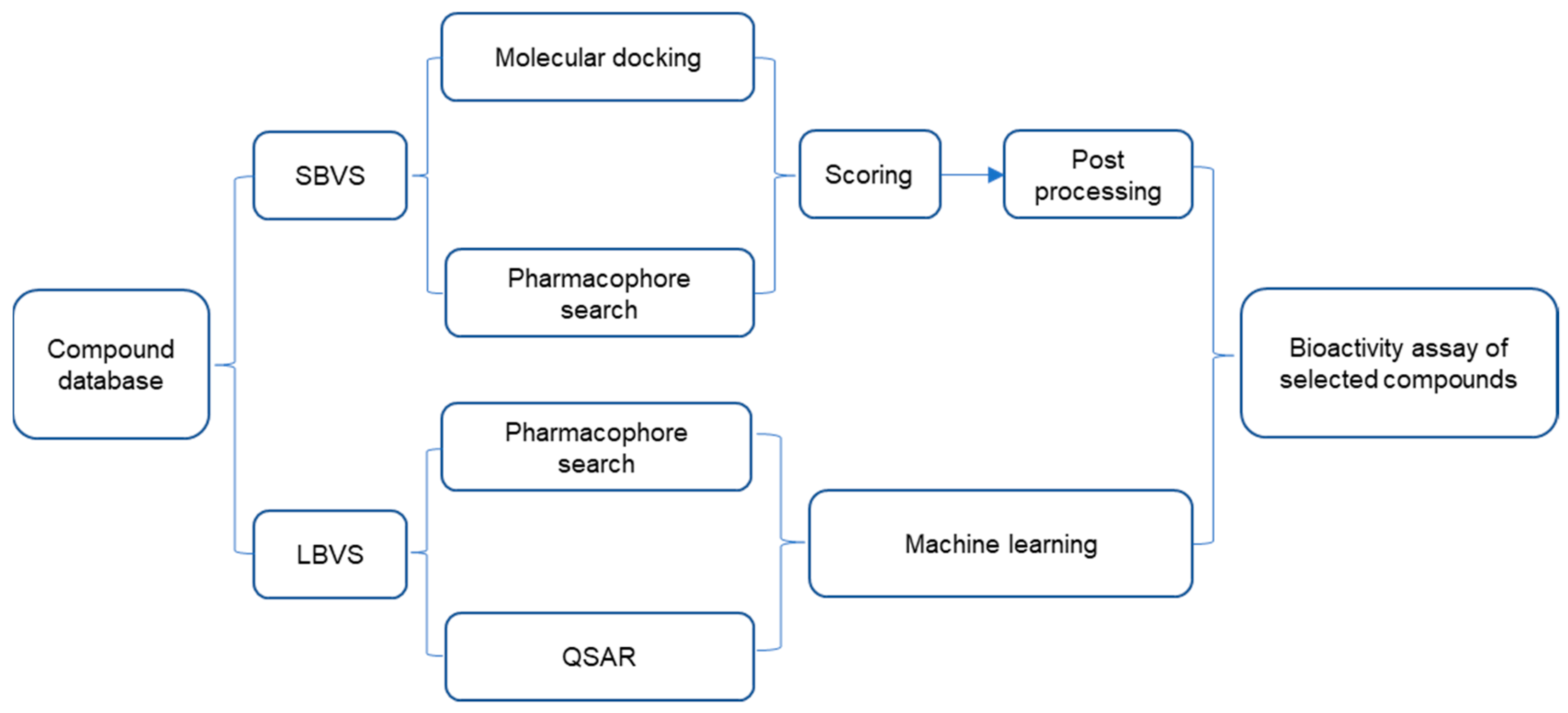
3. Strategies in the Screening of Nrf2–Keap1 PPI Inhibitors
3.1. Screening Nrf2–Keap1 PPI Inhibitors Based on HTS and VS
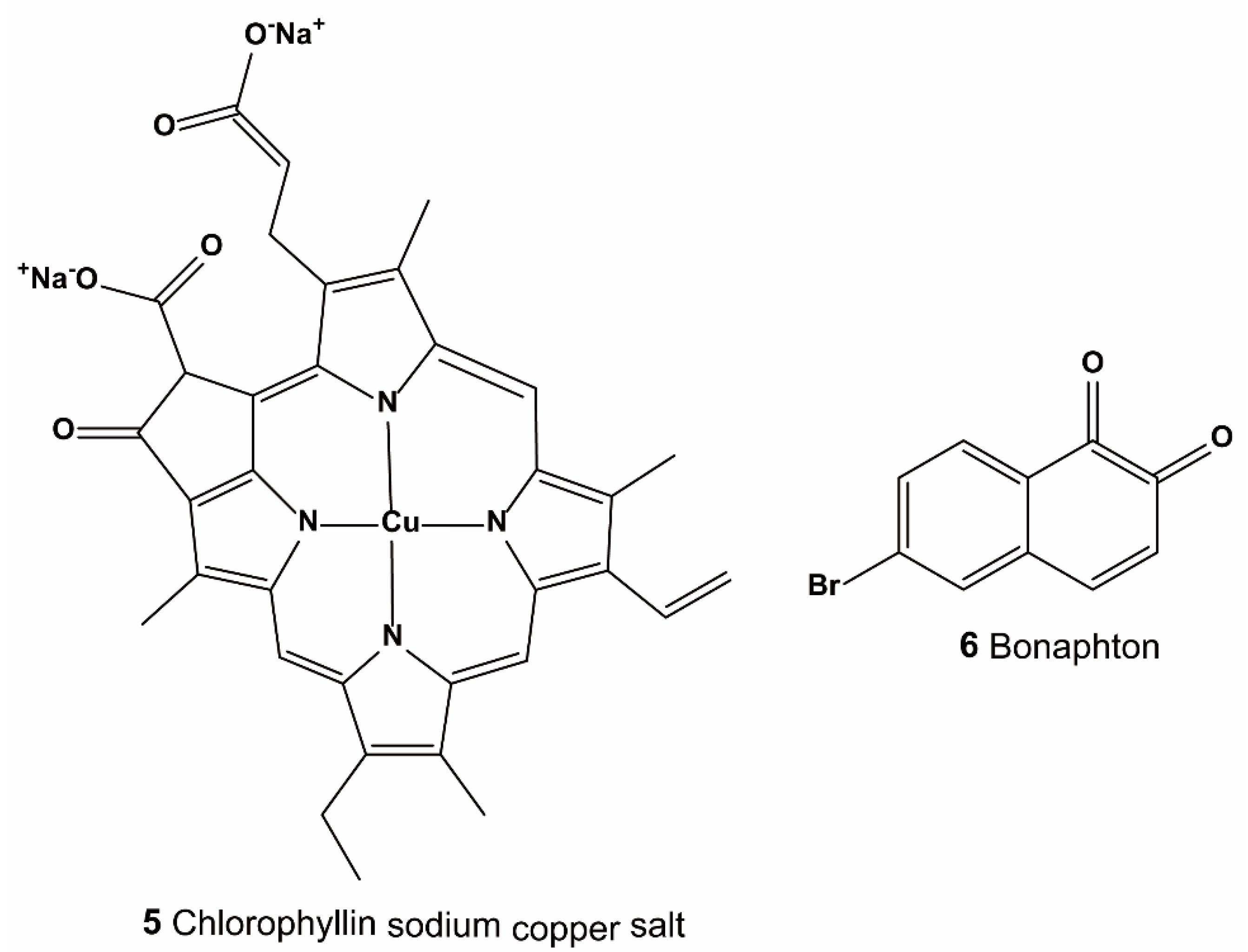
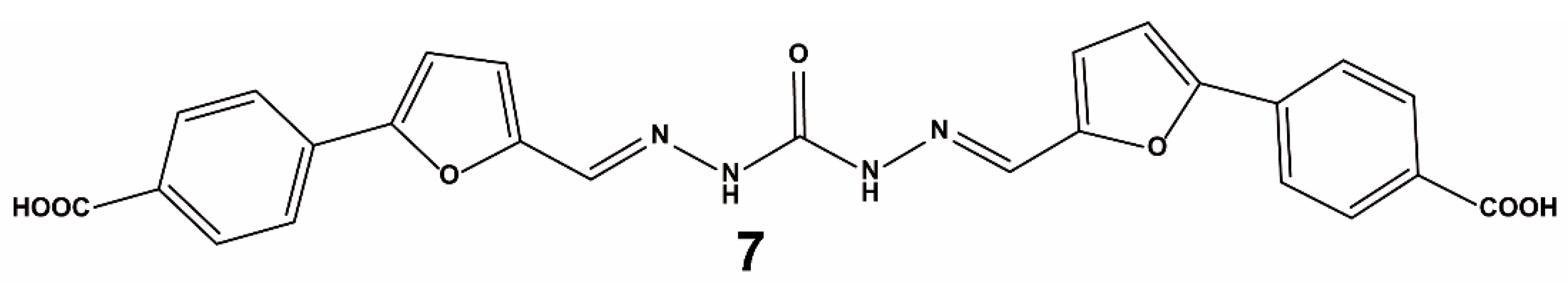
3.1.1. High-Throughput Screening of Nrf2–Keap1 Protein–Protein Interaction Inhibitors
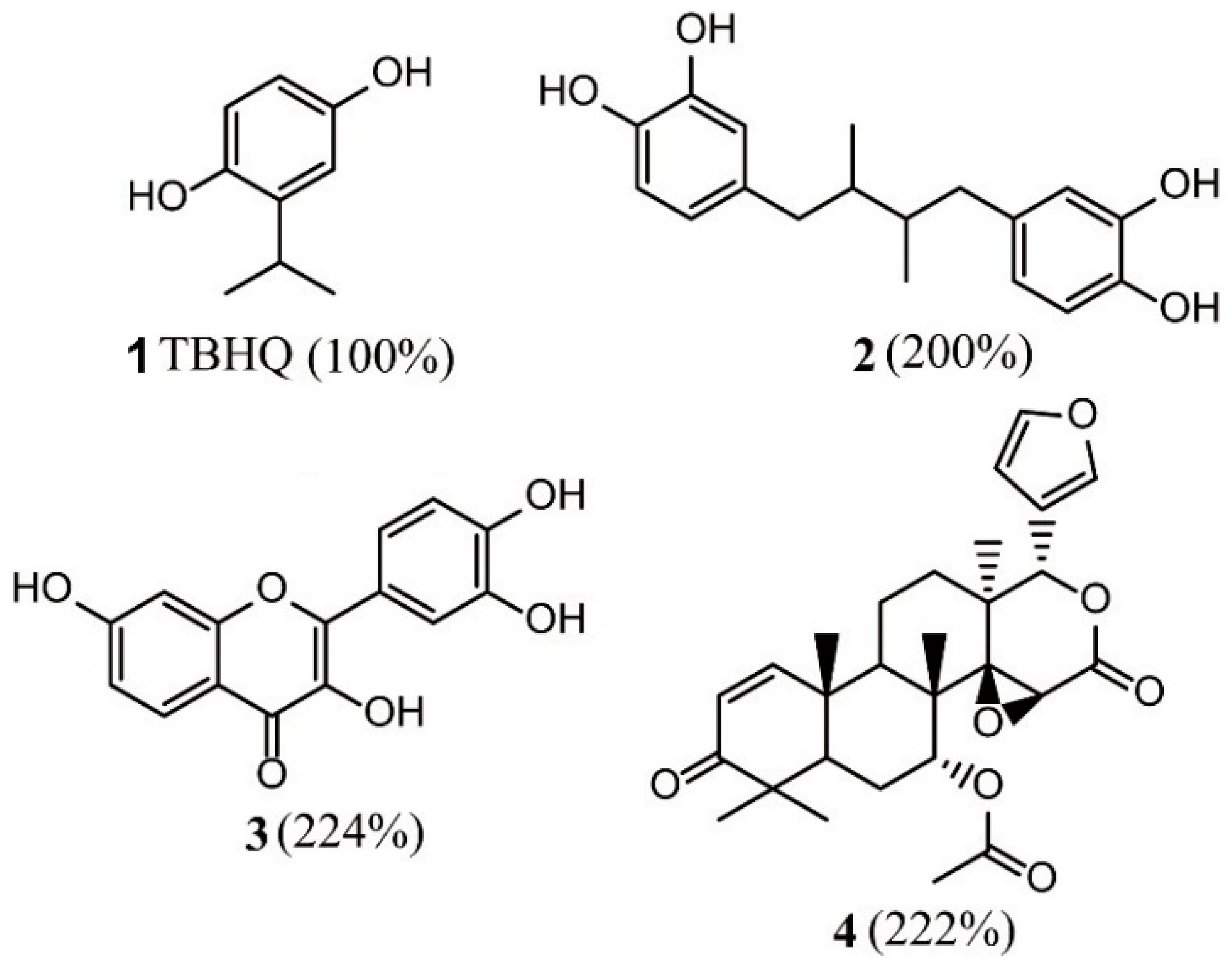
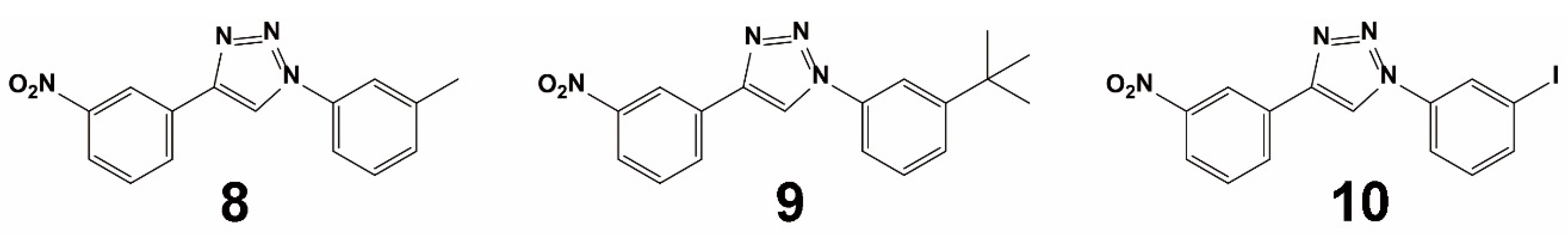
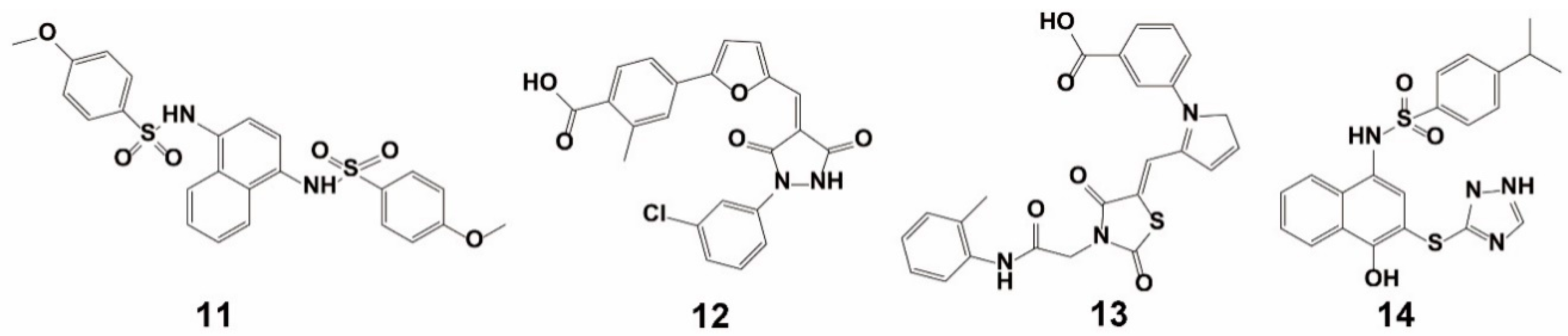
3.1.2. Virtual Screening
3.1.3. Combined Screening Using VS and HTS
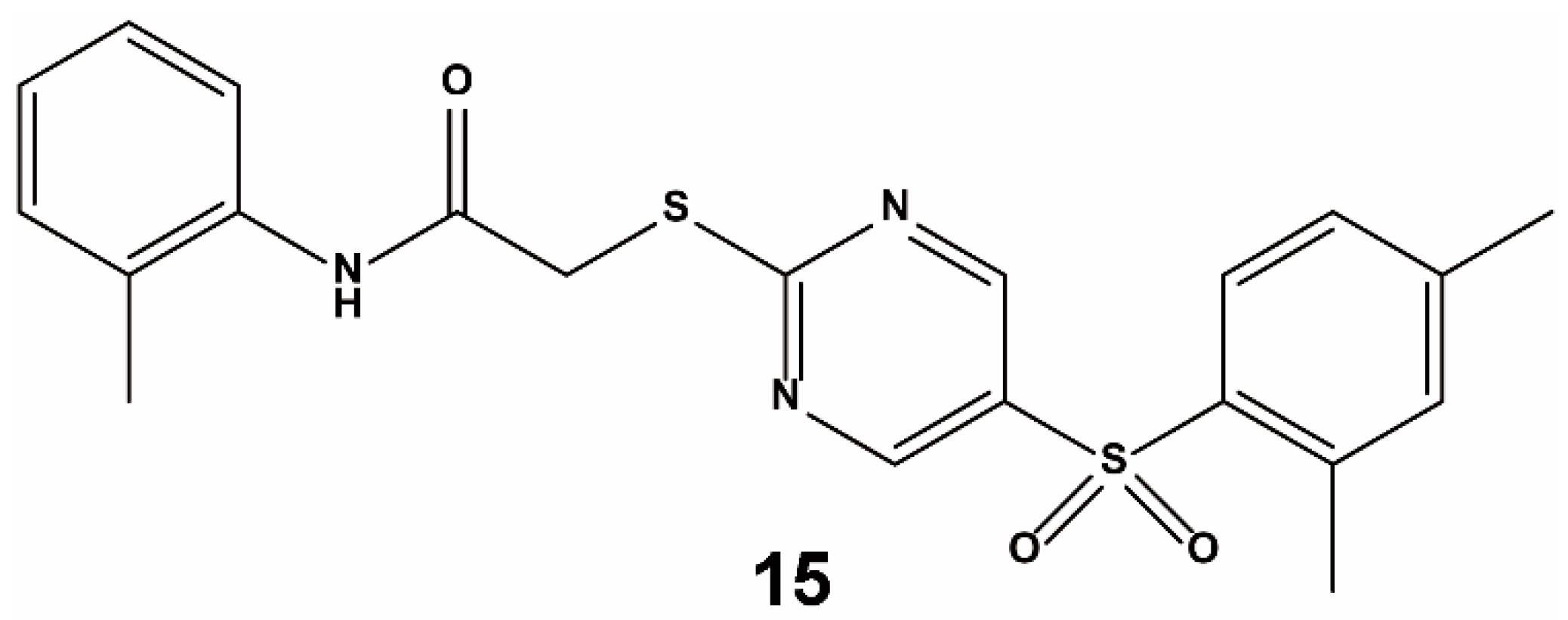
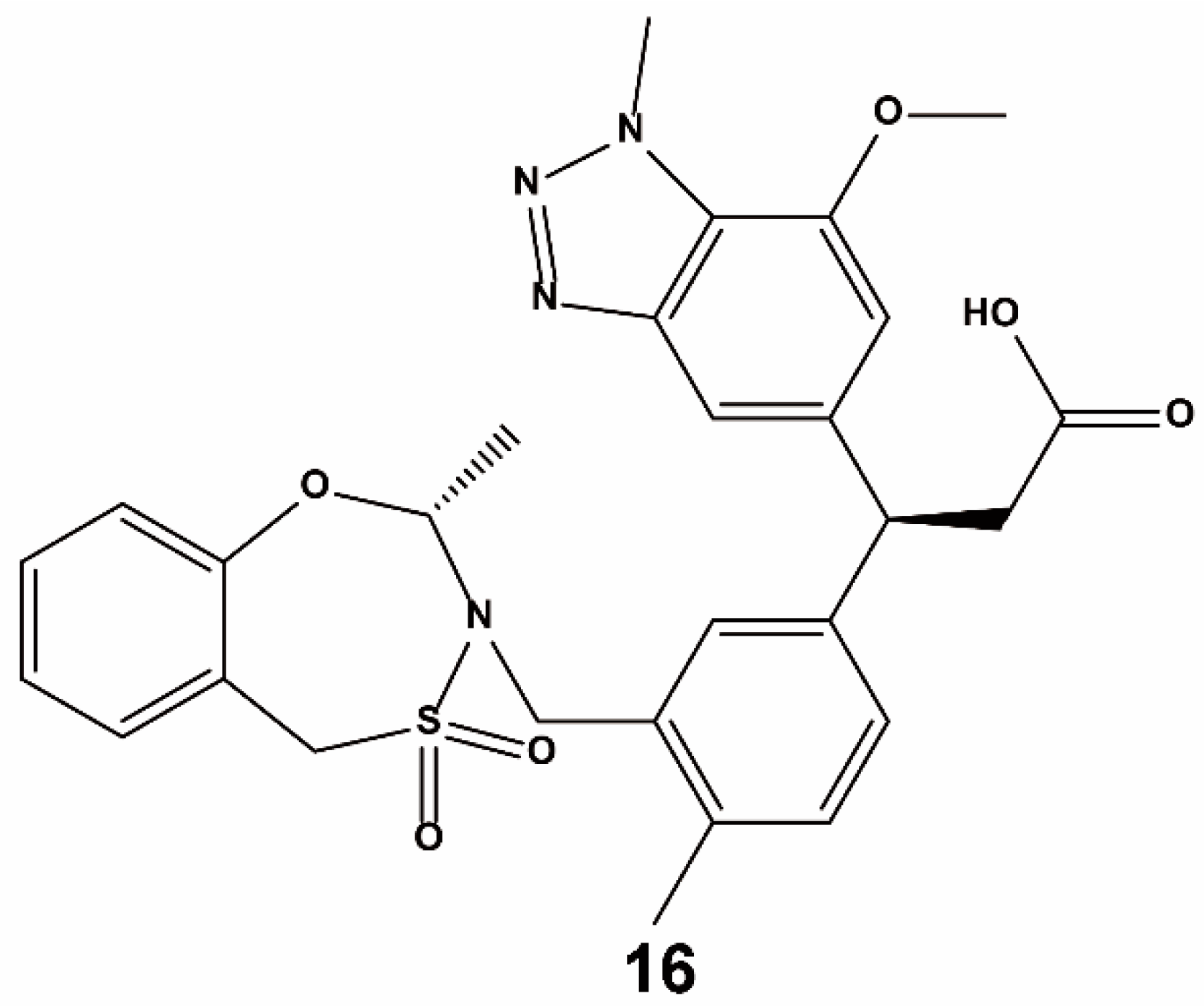
3.2. Fragment-Based Approach
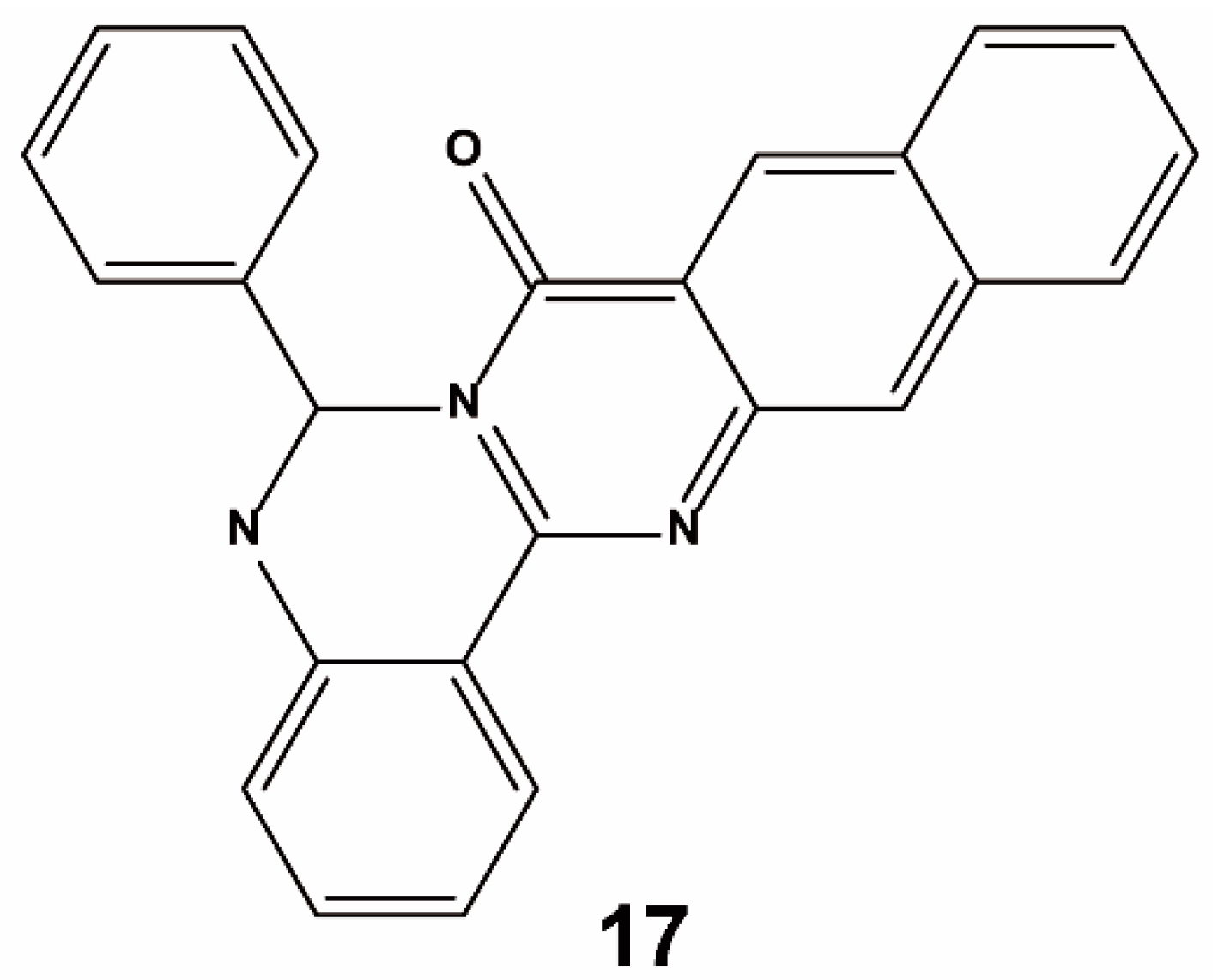
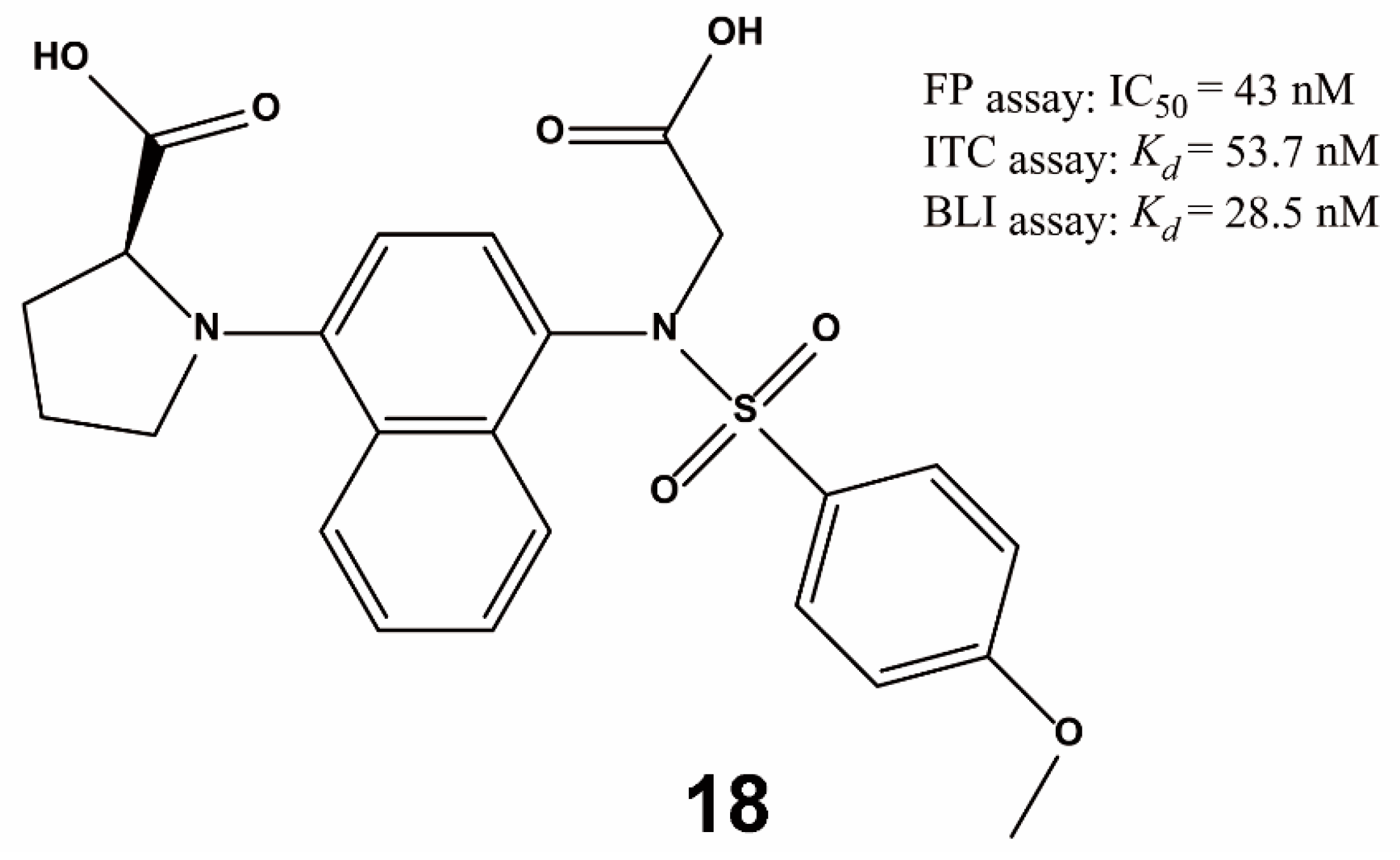
3.3. Others
4. Non-Screening Approaches for the Identification of Keap1–Nrf2 Interaction Inhibitors
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Meng, N.; Tang, H.; Zhang, H.; Jiang, C.; Su, L.; Min, X.; Zhang, W.; Zhang, H.; Miao, Z.; Zhang, W. Fragment-growing guided design of Keap1-Nrf2 protein-protein interaction inhibitors for targeting myocarditis. Free Radic. Biol. Med. 2018, 117, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Wu, Z.; Xing, C.; Miao, Z. Small molecules inhibiting Keap1-Nrf2 protein-protein interactions: A novel approach to activate Nrf2 function. Med. Chem. Comm. 2017, 8, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Chou, C.-W.; Kumar, K.S.; Fu, K.-T.; Wang, H.-M.; Hsu, L.-S.; Kuo, Y.-H.; Wu, C.-R.; Chen, S.-C.; Yang, H.-L. Ellagic acid protects human keratinocyte (HaCaT) cells against UVA-induced oxidative stress and apoptosis through the upregulation of the HO-1 and Nrf-2 antioxidant genes. Food Chem. Toxicol. 2012, 50, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Römer, S.; Boutten, A. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef]
- Hancock, R.; Schaap, M.; Pfister, H.; Wells, G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction with improved binding and cellular activity. Org. Biomol. Chem. 2013, 11, 3553–3557. [Google Scholar] [CrossRef]
- Sun, H.-P.; Jiang, Z.-Y.; Zhang, M.-Y.; Lu, M.-C.; Yang, T.-T.; Pan, Y.; Huang, H.-Z.; Zhang, X.-J.; You, Q.-D. Novel protein-protein interaction inhibitor of Nrf2-Keap1 discovered by structure-based virtual screening. MedChemComm 2014, 5, 93–98. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [PubMed]
- Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. Pharmacological Applications of Nrf2 Inhibitors as Potential Antineoplastic Drugs. Int. J. Mol. Sci. 2019, 20, 2025. [Google Scholar] [CrossRef] [PubMed]
- Abed, D.A.; Goldstein, M.; Albanyan, H.; Jin, H.; Hu, L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhang, B.; Di, J.; Jiang, G.; Chen, F.; Li, H.; Li, L.; Pei, D.; Zheng, J. Keap1: One stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett. 2012, 325, 26–34. [Google Scholar] [CrossRef]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Marzec, J.; Kleeberger, S.R. Functional polymorphisms in Nrf2: Implications for human disease. Free Radic. Biol. Med. 2015, 88, 362–372. [Google Scholar] [CrossRef]
- Pedruzzi, L.M.; Stockler-Pinto, M.B.; Leite, M., Jr.; Mafra, D. Nrf2–keap1 system versus NF-κB: The good and the evil in chronic kidney disease? Biochimie 2012, 94, 2461–2466. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Free Radic. Biol. Med. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Deck, L.M.; Hunsaker, L.A.; Vander Jagt, T.A.; Whalen, L.J.; Royer, R.E.; Vander Jagt, D.L. Activation of anti-oxidant Nrf2 signaling by enone analogues of curcumin. Eur. J. Med. Chem. 2018, 143, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Jiang, Z.; You, Q. Discovery and Development of Keap1-Nrf2 Protein-Protein Interaction Inhibitors. In Targeting Protein-Protein Interactions by Small Molecules; Springer: Berlin, Germany, 2018; pp. 249–286. [Google Scholar]
- Holland, R.; Navamal, M.; Velayutham, M.; Zweier, J.L.; Kensler, T.W.; Fishbein, J.C. Hydrogen peroxide is a second messenger in phase 2 enzyme induction by cancer chemopreventive dithiolethiones. Chem. Res. Toxicol. 2009, 22, 1427–1434. [Google Scholar] [CrossRef][Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Keum, Y.-S. NRF2, a key regulator of antioxidants with two faces towards cancer. Oxid. Med. Cell. Longev. 2016, 2016, 2746457. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kensler, T. Nrf2 as a target for cancer chemoprevention. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2005, 591, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2009, 31, 90–99. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Takahashi, H.; Jin, C.; Rajabi, H.; Pitroda, S.; Alam, M.; Ahmad, R.; Raina, D.; Hasegawa, M.; Suzuki, Y.; Tagde, A. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene 2015, 34, 5187. [Google Scholar] [CrossRef]
- Ramos-Gomez, M.; Kwak, M.-K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef]
- Osburn, W.O.; Karim, B.; Dolan, P.M.; Liu, G.; Yamamoto, M.; Huso, D.L.; Kensler, T.W. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer 2007, 121, 1883–1891. [Google Scholar] [CrossRef]
- Xu, C.; Huang, M.-T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.-N.T. Inhibition of 7, 12-dimethylbenz (a) anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2–related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodríguez-Oroz, M.C.; Benitez-Temino, B.; Blesa, F.J.; Guridi, J.; Marin, C.; Rodriguez, M. Functional organization of the basal ganglia: Therapeutic implications for Parkinson’s disease. Mov. Disord. 2008, 23, S548–S559. [Google Scholar] [CrossRef] [PubMed]
- Riedl, A.G.; Watts, P.M.; Brown, C.T.; Jenner, P. P450 and heme oxygenase enzymes in the basal ganglia and their roles in Parkinson’s disease. Adv. Neurol. 1999, 80, 271. [Google Scholar] [PubMed]
- Jakel, R.J.; Kern, J.T.; Johnson, D.A.; Johnson, J.A. Induction of the protective antioxidant response element pathway by 6-hydroxydopamine in vivo and in vitro. Toxicol. Sci. 2005, 87, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Magesh, S.; Chen, Y.; Hu, L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.I.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Osorio, A.; Picazo, A.; González-Reyes, S.; Barrera-Oviedo, D.; Rodríguez-Arellano, M.; Pedraza-Chaverri, J. Nrf2 and redox status in prediabetic and diabetic patients. Int. J. Mol. Sci. 2014, 15, 20290–20305. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.S.; Zhuang, C.L.; Zhu, K.; Zhang, J.; Muehlmann, L.A.; Figueiró Longo, J.P.; Azevedo, R.B.; Zhang, W.; Meng, N.; Zhang, H. Identification of a novel small-molecule Keap1-Nrf2 PPI inhibitor with cytoprotective effects on LPS-induced cardiomyopathy. J. Enzym. Inhib. Med. Chem. 2018, 33, 833–884. [Google Scholar] [CrossRef]
- Huang, L.; Wang, J.; Chen, L.; Zhu, M.; Wu, S.; Chu, S.; Zheng, Y.; Fan, Z.; Zhang, J.; Li, W.; et al. Design, synthesis, and evaluation of NDGA analogues as potential anti-ischemic stroke agents. Eur. J. Med. Chem. 2018, 143, 1165–1173. [Google Scholar] [CrossRef]
- Choi, S.H.; Park, S.; Oh, C., Jr.; Leem, J.; Park, K.G.; Lee, I.K. Dipeptidyl peptidase-4 inhibition by gemigliptin prevents abnormal vascular remodeling via NF-E2-related factor 2 activation. Vascul. Pharmacol. 2015, 73, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ichikawa, T.; Janicki, J.S.; Cui, T. Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin. Ther. Targets 2009, 13, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Bioph. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Wanner, J.; Fry, D.C.; Peng, Z.; Roberts, J. Druggability assessment of protein–protein interfaces. Future Med. Chem. 2011, 3, 2021–2038. [Google Scholar] [CrossRef]
- Smirnova, N.A.; Haskew-Layton, R.E.; Basso, M.; Hushpulian, D.M.; Payappilly, J.B.; Speer, R.E.; Ahn, Y.-H.; Rakhman, I.; Cole, P.A.; Pinto, J.T. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem. Biol. 2011, 18, 752–765. [Google Scholar] [CrossRef]
- Yoshizaki, Y.; Mori, T.; Ishigami-Yuasa, M.; Kikuchi, E.; Takahashi, D.; Zeniya, M.; Nomura, N.; Mori, Y.; Araki, Y.; Ando, F.; et al. Drug-Repositioning Screening for Keap1-Nrf2 Binding Inhibitors using Fluorescence Correlation Spectroscopy. Sci. Rep. 2017, 7, 3945. [Google Scholar] [CrossRef]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1–Nrf2 protein–protein interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.; Zeng, W.; Hus, J.-C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorgan. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.A.; Cohen, D.S.; Jarrell, J.T.; Huang, X. Deep learning and virtual drug screening. Future Med. Chem. 2018, 10, 2557–2567. [Google Scholar] [CrossRef]
- Manoharan, P.; Ghoshal, N. Fragment-based virtual screening approach and molecular dynamics simulation studies for identification of BACE1 inhibitor leads. J Biomol. Struct. Dyn. 2018, 36, 1878–1892. [Google Scholar] [CrossRef]
- Lengauer, T.; Lemmen, C.; Rarey, M.; Zimmermann, M. Novel technologies for virtual screening. Drug Discov. Today 2004, 9, 27–34. [Google Scholar] [CrossRef]
- Shi, M.; Xu, D.; Zeng, J. GPU Accelerated Quantum Virtual Screening: Application for the Natural Inhibitors of New Dehli Metalloprotein (NDM-1). Front. Chem. 2018, 6, 564. [Google Scholar] [CrossRef]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Cuozzo, J. High-throughput affinity-based technologies for small-molecule drug discovery. J. Biomol. Screen. 2009, 14, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Cavasotto, C.N. Open challenges in structure-based virtual screening: Receptor modeling, target flexibility consideration and active site water molecules description. Arch. Biochem. Biophys. 2015, 583, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935. [Google Scholar] [CrossRef] [PubMed]
- Heightman, T.D.; Callahan, J.F.; Chiarparin, E.; Coyle, J.E.; Griffiths-Jones, C.; Lakdawala, A.S.; McMenamin, R.; Mortenson, P.N.; Norton, D.; Peakman, T.M.; et al. Structure-Activity and Structure-Conformation Relationships of Aryl Propionic Acid Inhibitors of the Kelch-like ECH-Associated Protein 1/Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1/NRF2) Protein-Protein Interaction. J. Med. Chem. 2019, 62, 4683–4702. [Google Scholar] [CrossRef] [PubMed]
- Schaap, M.; Hancock, R.; Wilderspin, A.; Wells, G. Development of a steady-state FRET-based assay to identify inhibitors of the Keap1-Nrf2 protein-protein interaction. Protein Sci. 2013, 22, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, X.; Wang, G.; Barbour, K.W.; Berger, F.G.; Wang, Q. Drug screening assay based on the interaction of intact Keap1 and Nrf2 proteins in cancer cells. Bioorg. Med. Chem. 2019, 27, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.; Bertrand, H.C.; Tsujita, T.; Naz, S.; El-Bakry, A.; Laoruchupong, J.; Hayes, J.D.; Wells, G. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic. Biol. Med. 2012, 52, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Alsaid, M.S.; El-Gazzar, M.G.; Higgins, M.; Dinkova-Kostova, A.T.; Shahat, A.A. Synthesis and biological evaluation of novel 2-phenylquinazoline-4-amine derivatives: Identification of 6-phenyl-8H-benzo[g]quinazolino[4,3-b]quinazolin-8-one as a highly potent inducer of NAD (P) H quinone oxidoreductase 1. J. Enzym. Inhib. Med. Chem. 2016, 31, 34–39. [Google Scholar] [CrossRef]
- Lu, M.C.; Zhang, X.; Wu, F.; Lu, M.C.; Zhao, J.; You, Q.D.; Jiang, Z.Y. Discovery of a Potent Kelch-Like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Protein-Protein Interaction Inhibitor with Natural Proline Structure as a Cytoprotective Agent against Acetaminophen-Induced Hepatotoxicity. J. Med. Chem. 2019, 62, 6796–6813. [Google Scholar] [CrossRef]
- Lu, M.C.; Jiao, Q.; Liu, T.; Tan, S.J.; Zhou, H.S.; You, Q.D.; Jiang, Z.Y. Discovery of a head-to-tail cyclic peptide as the Keap1-Nrf2 protein-protein interaction inhibitor with high cell potency. Eur. J. Med. Chem. 2018, 143, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
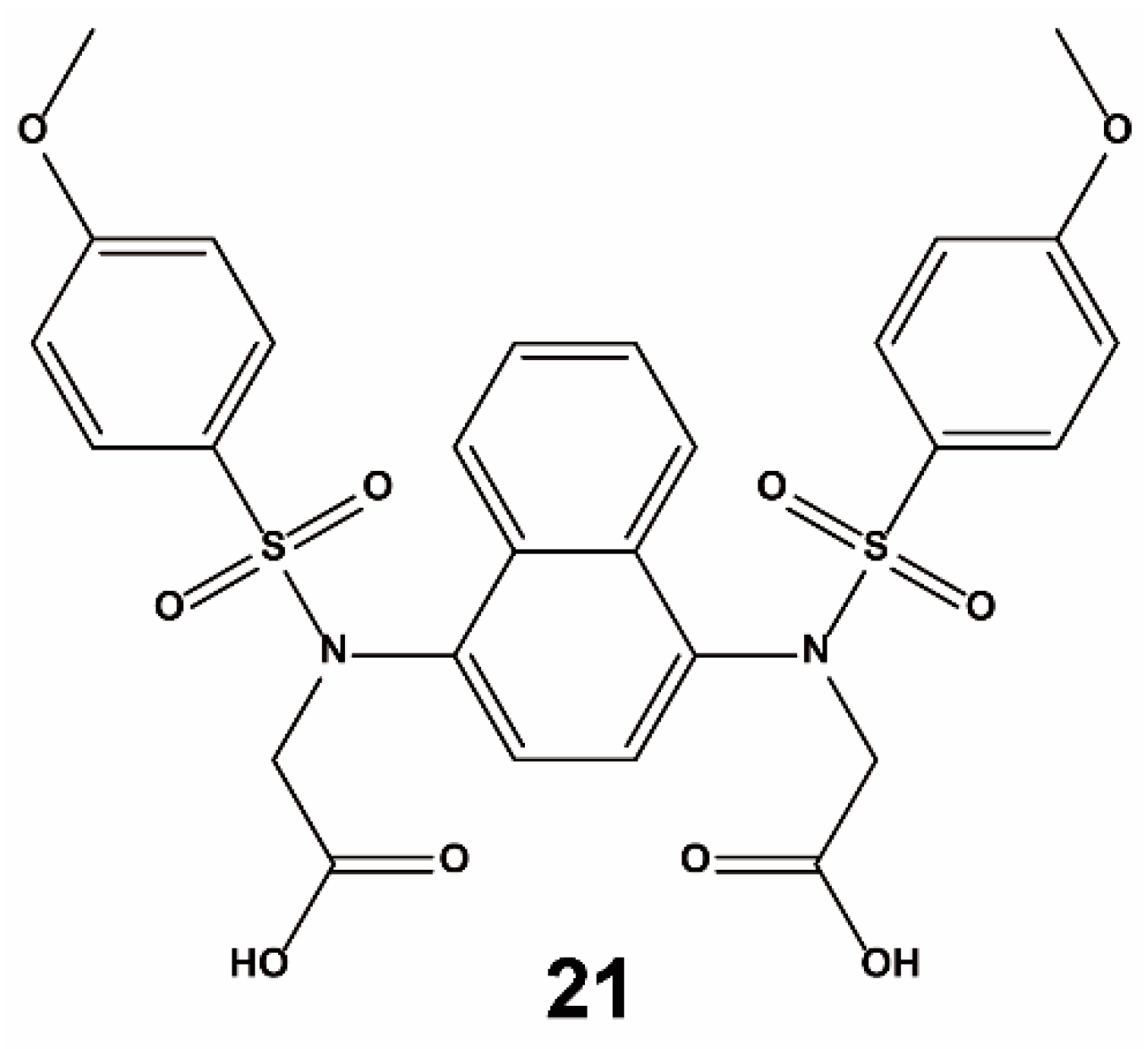
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Poore, D.D.; Hofmann, G.; Wolfe, L.A.; Qi, H.; Jiang, M.; Fischer, M.; Wu, Z.; Sweitzer, T.D.; Chakravorty, S.; Donovan, B.; et al. Development of a High-Throughput Cul3-Keap1 Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Assay for Identifying Nrf2 Activators. SLAS Discov. 2019, 24, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Wells, G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 2015, 43, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hou, Y.; Liu, C.; Li, Y.; Guo, W.; Wu, J.-L.; Xu, D.; You, X.; Pan, Y.; Chen, Y. Identification of an adaptor protein that facilitates Nrf2-Keap1 complex formation and modulates antioxidant response. Free Radic. Biol. Med. 2016, 97, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Waller, C.L.; Swaan, P.W.; Cruciani, G.; Wrighton, S.A.; Wikel, J.H. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods 2000, 44, 251–272. [Google Scholar] [CrossRef]
- Yang, G.J.; Lei, P.M.; Wong, S.Y.; Ma, D.L.; Leung, C.H. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules 2018, 23, E3194. [Google Scholar] [CrossRef]
- Lu, M.C.; Zhao, J.; Liu, Y.T.; Liu, T.; Tao, M.M.; You, Q.D.; Jiang, Z.Y. CPUY192018, a potent inhibitor of the Keap1-Nrf2 protein-protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF-κB activation. Redox Biol. 2019, 26, 101266. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Xu, L.L.; Lu, M.C.; Chen, Z.Y.; Yuan, Z.W.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; Sun, H.P.; You, Q.D. Structure-Activity and Structure-Property Relationship and Exploratory In Vivo Evaluation of the Nanomolar Keap1-Nrf2 Protein-Protein Interaction Inhibitor. J. Med. Chem. 2015, 58, 6410–6421. [Google Scholar] [CrossRef]
- Quinti, L.; Dayalan Naidu, S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef]
- Feng, S.; Qiu, B.; Zou, L.; Liu, K.; Xu, X.; Zhu, H. Schisandrin B elicits the Keap1-Nrf2 defense system via carbene reactive metabolite which is less harmful to mice liver. Drug Des. Devel. Ther. 2018, 12, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.L.; Zhang, J.; Liu, X.; Gao, J.Y. Role of growth hormone in maturation and activation of dendritic cells via miR-200a and the Keap1/Nrf2 pathway. Cell prolif. 2015, 48, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Lnher-Melville, K.1.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jian, T.; Wu, Y.; Zuo, Y.; Li, J.; Lv, H.; Ma, L.; Ren, B.; Zhao, L.; Li, W.; et al. Ellagic acid ameliorates oxidative stress and insulin resistance in high glucose-treated HepG2 cells via miR-223/keap1-Nrf2 pathway. Biomed. Pharmacother. 2019, 110, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Chen, F.; Lv, H.; Xu, Z.; Fu, J.; Hou, Y.; Xu, Y.; Pi, J. Triptolide enhances chemotherapeutic efficacy of antitumor drugs in non-small-cell lung cancer cells by inhibiting Nrf2-ARE activity. Toxicol. Appl. Pharmacol. 2018, 358, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Gross-Steinmeyer, K.; Eaton, D.L. Dietary modulation of the biotransformation and genotoxicity of aflatoxin B(1). Toxicology 2012, 299, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Corna, D.; Nava, V.; Locatelli, M.; Abbate, M.; Gaspari, F.; Carrara, F.; Sangalli, F.; Remuzzi, G.; Benigni, A. Analogs of bardoxolone methyl worsen diabetic nephropathy in rats with additional adverse effects. Am. J. Physiol. Renal. Physiol. 2013, 304, F808–F819. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cochemé, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
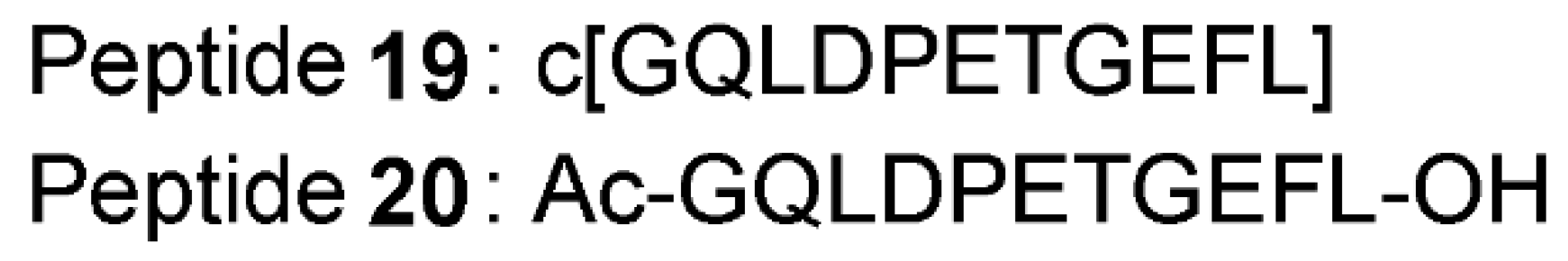{kind=link}
{kind=link}
| Method | Library Size | Screening Software | Ref |
|---|---|---|---|
| High-throughput screening (HTS) | 2000 compounds | - | [60] |
| 1633 drugs | - | [61] | |
| Virtual screening (VS) | 251,774 compounds | Ligandfit | [10] |
| ~178,000 compounds | Autodock 4.2 and DOCK 6.6 | [62] | |
| HTS + VS | (267,551 + 1911 compounds) | Glide 5.5 | [63] |
| 300,000 compounds | Schrodinger’s Glide | [64] | |
| Fragment-based approach | 330 molecular fragments | - | [65] |
| Methods | Benefits | Drawbacks |
|---|---|---|
| VS | Low cost; high-throughput | High false positive rate; only applicable for primary screening |
| SPR a | Label-free; low false positive rate | Low-throughput; high technical and equipment requirements; high cost |
| ITC | ||
| BLI | ||
| BIFC-based assay [79] b | High-throughput; label-free; low false positive rate | Low sensitivity |
| ARE gene promoter assay [61] Phage peptide display [79] c | Low sensitivity | |
| FP-based assay [79] | High-throughput, easy to operate; high sensitivity | Labeling requirement; interference by compounds with autofluorescence or fluorescence quenching ability |
| FCS-based assay [61] d TR-FRET-based assay [77,84] e |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, C.-H.; Zhang, J.-T.; Yang, G.-J.; Liu, H.; Han, Q.-B.; Ma, D.-L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445. https://doi.org/10.3390/ijms20184445
Leung C-H, Zhang J-T, Yang G-J, Liu H, Han Q-B, Ma D-L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. International Journal of Molecular Sciences. 2019; 20(18):4445. https://doi.org/10.3390/ijms20184445
Chicago/Turabian StyleLeung, Chung-Hang, Jia-Tong Zhang, Guan-Jun Yang, Hao Liu, Quan-Bin Han, and Dik-Lung Ma. 2019. "Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors" International Journal of Molecular Sciences 20, no. 18: 4445. https://doi.org/10.3390/ijms20184445
APA StyleLeung, C.-H., Zhang, J.-T., Yang, G.-J., Liu, H., Han, Q.-B., & Ma, D.-L. (2019). Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. International Journal of Molecular Sciences, 20(18), 4445. https://doi.org/10.3390/ijms20184445