Lysine Acetylation is an Important Post-Translational Modification that Modulates Heat Shock Response in the Sea Cucumber Apostichopus japonicus


Abstract
1. Introduction
2. Results
2.1. A. japonicus Had a Large Number of Acetylated Proteins and Sites by Proteome-Wide Analysis
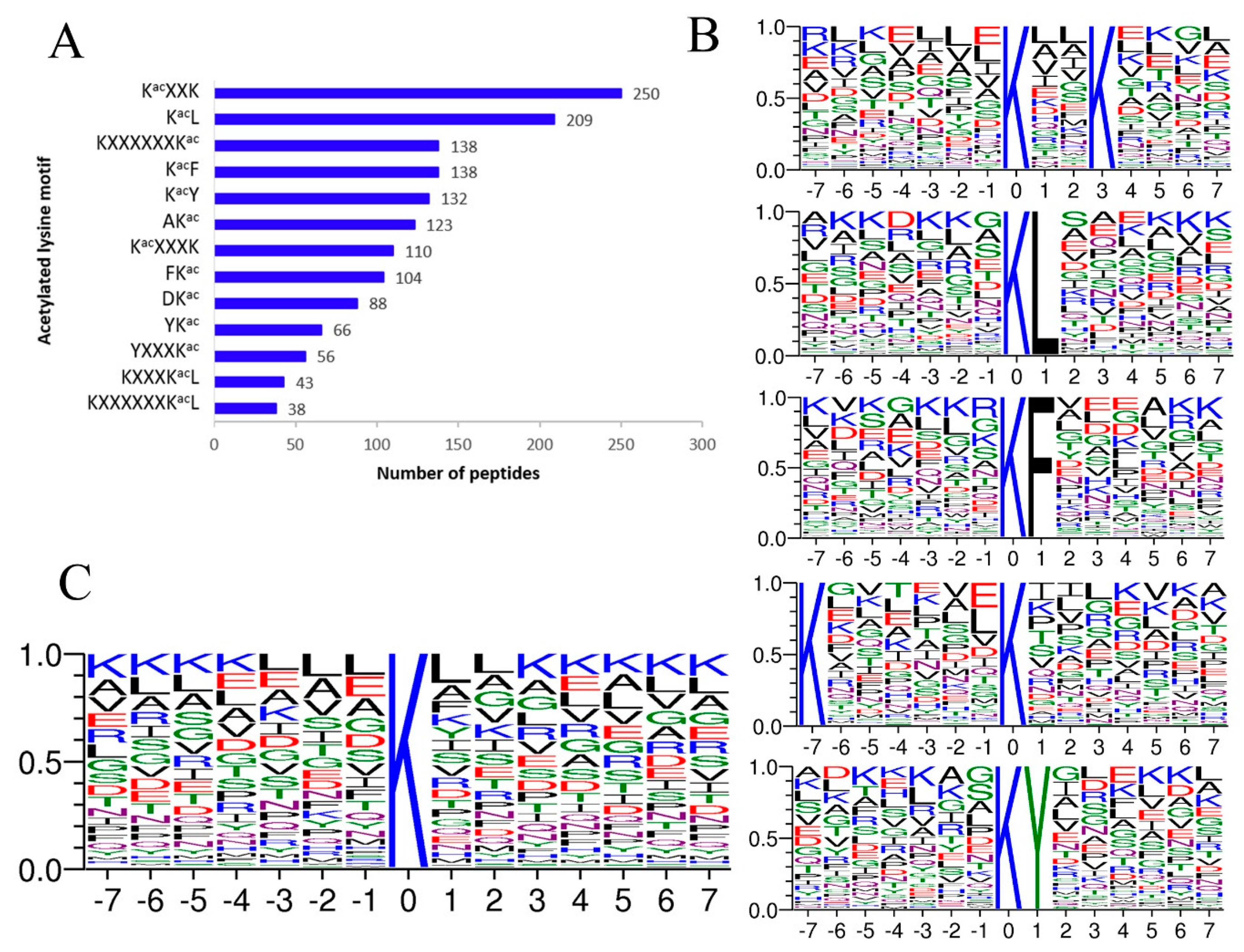
2.2. 13 Acetylation Motifs Were Characterized in A. japonicus
2.3. Functional Annotation of Acetylated Proteins
2.4. The Number of Acetylation Sites in Many Functional Proteins Changed Significantly under Heat Stress (HS)
2.4.1. HS6h versus C Comparison
2.4.2. HS48h versus C Comparison
2.4.3. HS48h versus HS6h Comparison
2.5. Differentially Acetylated Proteins Represented Specific Interaction Networks
3. Discussion
3.1. Lysine Acetylation Profiles in A. japonicus
3.2. Differential Number of Acetylated Sites under HS
3.2.1. Lysine Acetyltransferases (KATs) and Deacetylases (KDACs)
3.2.2. Chaperones
3.2.3. Translation-Associated Factors and Ribosome Proteins
4. Materials and Methods
4.1. Samples
4.2. Protein Extraction
4.3. Trypsin Digestion and ENRICHMENT of Lysine-Acetylated Peptides
4.4. Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) Detection and Data Analysis
4.5. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hashiguchi, A.; Komatsu, S. Impact of Post-Translational Modifications of Crop Proteins under Abiotic Stress. Proteomes 2016, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Dikic, I.; Zhou, M.-M.; Pawson, T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.; Faulkner, R.; Mirsky, A. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Histone acetylation and an epigenetic code. Bioessays 2000, 22, 836–845. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, L.; Liang, W.; Mu, P.; Wang, S.; Lin, Q. Comprehensive profiling of lysine acetylproteome analysis reveals diverse functions of lysine acetylation in common wheat. Sci. Rep. 2016, 6, 21069. [Google Scholar] [CrossRef]
- Tauber, C.A.; Tauber, M.J. Insect seasonal cycles: Genetics and evolution. Annu. Rev. Ecol. Syst. 1981, 12, 281–308. [Google Scholar] [CrossRef]
- Hentchel, K.L.; Escalante-Semerena, J.C. Acylation of biomolecules in prokaryotes: A widespread strategy for the control of biological function and metabolic stress. Microbiol. Mol. Biol. Rev. 2015, 79, 321–346. [Google Scholar] [CrossRef]
- Neilson, K.A.; Gammulla, C.G.; Mirzaei, M.; Imin, N.; Haynes, P.A. Proteomic analysis of temperature stress in plants. Proteomics 2010, 10, 828–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, M.; Wang, X.; Yang, S.; Gu, J.; Zhou, J.; Zhang, X.-E.; Deng, J.; Ge, F. Acetylome analysis reveals diverse functions of lysine acetylation in Mycobacterium tuberculosis. Mol. Cell. Proteom. 2014, 13, 3352–3366. [Google Scholar]
- Ma, Q.; Wood, T.K. Protein acetylation in prokaryotes increases stress resistance. Biochem. Biophys. Res. Commun. 2011, 410, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Ozden, O.; Park, S.-H.; Kim, H.-S.; Jiang, H.; Coleman, M.C.; Spitz, D.R.; Gius, D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging 2011, 3, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Bao, Z.; Hou, R.; Wang, S.; Su, H.; Yan, J.; Tian, M.; Li, Y.; Wei, W.; Lu, W.; et al. Transcriptome Sequencing and Characterization for the Sea Cucumber Apostichopus japonicus (Selenka, 1867). PLoS ONE 2012, 7, e33311. [Google Scholar] [CrossRef] [PubMed]
- Hoegh-Guldberg, O.; Bruno, J.F. The impact of climate change on the world’s marine ecosystems. Science 2010, 328, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fang, H.; Xu, D. Effect of seasonal high temperature on the immune response in Apostichopus japonicus by transcriptome analysis. Fish Shellfish Immunol. 2019, 92, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sun, L.; Liu, S.; Zhang, L.; Ru, X.; Zhao, Y.; Yang, H. Molecular cloning of heat shock protein 10 (Hsp10) and 60 (Hsp60) cDNAs and their expression analysis under thermal stress in the sea cucumber Apostichopus japonicus. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014, 171, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, H.; Zhao, H.; Chen, M.; Wang, T. The molecular characterization and expression of heat shock protein 90 (Hsp90) and 26 (Hsp26) cDNAs in sea cucumber (Apostichopus japonicus). Cell Stress Chaperones 2011, 16, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhou, S.; Sun, L. RNA-seq based transcriptional analysis reveals dynamic genes expression profiles and immune-associated regulation under heat stress in Apostichopus japonicus. Fish Shellfish Immunol. 2018, 78, 168–176. [Google Scholar] [CrossRef]
- Schwartz, D.; Chou, M.F.; Church, G.M. Predicting Protein Post-translational Modifications Using Meta-analysis of Proteome Scale Data Sets. Mol. Cell Proteom. 2009, 8, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, Z.-H.; You, D.; Zhou, Y.; Ye, B.-C. Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea. Appl. Microbiol. Biotechnol. 2015, 99, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Shen, H.; Zhang, H.; Yan, J.; Liu, Y.; Yao, F.; Wang, X.; Cheng, Z.; Tang, T.-S.; Guo, C. Quantitative proteomics analysis reveals alterations of lysine acetylation in mouse testis in response to heat shock and X-ray exposure. BBA Proteins Proteom. 2018, 1866, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Gai, Z.; Wang, Y.; Fan, K.; Sun, L.; Wang, H.; Ding, Z. Comprehensive proteome analyses of lysine acetylation in tea leaves by sensing nitrogen nutrition. BMC Genom. 2018, 19, 840. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-R.; Yan, X.; Zhu, D.; Deng, X.; Wu, J.-S.; Xia, J.; Yan, Y.-M. Lysine acetylproteome profiling under water deficit reveals key acetylated proteins involved in wheat grain development and starch biosynthesis. J. Proteom. 2018, 185, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, Y.; Jin, M.; He, J.; Guli, A.; Yan, C.; Ding, S. Comprehensive analysis of lysine acetylome reveals a site-specific pattern in rapamycin-induced autophagy. J. Proteome Res. 2019, 18, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sun, L.; Liu, S.; Zhang, L.; Yang, H. Understanding the heat shock response in the sea cucumber Apostichopus japonicus, using iTRAQ-Based Proteomics. Int. J. Mol. Sci. 2016, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, L.; Yuan, J.; Sun, Y.; Gao, Y.; Zhang, L.; Li, S.; Dai, H.; Hamel, J.-F.; Liu, C.; et al. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 2017, 15, e2003790. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res. 2007, 17, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.R.; Wang, D.; Wang, L.; Fulco, M.; Pediconi, N.; Zhang, D.; An, W.; Ge, Q.; Roeder, R.G.; Wong, J.; et al. Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 2004, 11, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90 a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Matthias, P.; Yoshida, M.; Khochbin, S. HDAC6 a new cellular stress surveillance factor. Cell Cycle 2008, 7, 7–10. [Google Scholar] [CrossRef]
- Lee, J.B.; Wei, J.; Liu, W.; Cheng, J.; Feng, J.; Yan, Z. Histone deacetylase 6 gates the synaptic action of acute stress in prefrontal cortex. J. Physiol. 2012, 590, 1535–1546. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef]
- Fanghänel, J.; Fischer, G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci. 2004, 9, 3453–3478. [Google Scholar] [CrossRef]
- Sykes, K.; Gething, M.J.; Sambrook, J. Proline isomerases function during heat shock. Proc. Natl. Acad. Sci. USA 1993, 90, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Wek, R.C. Eukaryotic Initiation Factor 2 Phosphorylation and Translational Control in Metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Soufi, B.; Soares, N.C.; Ravikumar, V.; Macek, B. Proteomics reveals evidence of cross-talk between protein modifications in bacteria: Focus on acetylation and phosphorylation. Curr. Opin. Microbiol. 2012, 15, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Kamita, M.; Kimura, Y.; Ino, Y.; Kamp, R.M.; Polevoda, B.; Sherman, F.; Hirano, H. Nα-Acetylation of yeast ribosomal proteins and its effect on protein synthesis. J. Proteom. 2011, 74, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cimen, H.; Han, M.-J.; Shi, T.; Deng, J.-H.; Koc, H.; Palacios, O.M.; Montier, L.; Bai, Y.; Tong, Q. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J. Biol. Chem. 2010, 285, 7417–7429. [Google Scholar] [CrossRef]
- Xu, D.; Zhou, S.; Yang, H. Carbohydrate and amino acids metabolic response to heat stress in the intestine of the sea cucumber Apostichopus japonicus. Aquac. Res. 2017, 48, 5883–5891. [Google Scholar] [CrossRef]
- Liao, G.; Xie, L.; Li, X.; Cheng, Z.; Xie, J. Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J. Proteom. 2014, 106, 260–269. [Google Scholar] [CrossRef]
- Sun, L.; Lin, C.; Li, X.; Xing, L.; Huo, D.; Sun, J.; Zhang, L.; Yang, H. Comparative phospho-and acetyl proteomics analysis of posttranslational modifications regulating intestine regeneration in sea cucumbers. Front. Physiol. 2018, 9, 836. [Google Scholar] [CrossRef]
- Xu, Y.-X.; Chen, W.; Ma, C.-L.; Shen, S.-Y.; Zhou, Y.-Y.; Zhou, L.-Q.; Chen, L. Proteome and acetyl-proteome profiling of Camellia sinensis cv. ‘Anjin Baicha’ during periodic albinism reveals alterations in photosynthetic and secondary metabolite biosynthetic pathways. Front. Plant Sci. 2017, 8, 2104. [Google Scholar] [CrossRef]
- Fu, L.; Xu, Y.; Hou, Y.; Qi, X.; Zhou, L.; Liu, H.; Luan, Y.; Jing, L.; Miao, Y.; Zhao, S.; et al. Proteomic analysis indicates that mitochondrial energy metabolism in skeletal muscle tissue is negatively correlated with feed efficiency in pigs. Sci. Rep. 2017, 7, 45291. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Birney, E.; Biswas, M.; Bucher, P.; Cerutti, L.; Corpet, F.; Croning, M.D.R. InterPro—An integrated documentation resource for protein families, domains and functional sites. Bioinformatics 2000, 16, 1145–1150. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Function | Protein Name | Short Name | Acetylated Site | log2value |
|---|---|---|---|---|---|
| AJAP27758 | KAT KADC | histone acetyltransferase KAT5 | KAT5 | K124↑ | 0.86 |
| AJAP07562 | KAT KADC | KAT8 regulatory NSL complex subunit 1 | KANSL1 | K672↑ | 0.62 |
| AJAP05305 | KAT KADC | CREB-binding protein | CBP | K1804↑ | 0.46 |
| AJAP01782 | chromosomal stability | Fanconi anemia group J protein homolog | FANCJ | K274↑ | 1.28 |
| AJAP09062 | ribosome | large subunit ribosomal protein L3e | Ribosomal_L3e | K456↑ | 1.19 |
| AJAP03812 | ribosome | small subunit ribosomal protein S15e | Ribosomal_S15e | K78↑ | 1.00 |
| AJAP22222 | protein biosynthesis | translation initiation factor 2 subunit 1 | IF2 | K178↑ | 1.03 |
| AJAP07581 | protein biosynthesis | translation elongation factor 2 | EF2 | K19↑ | 0.67 |
| AJAP06040 | protein biosynthesis | translation elongation factor EF-1alpha (GTPase) | EF1α | K320↑ | 1.57 |
| AJAP00690 | protein binding | bromodomain adjacent to zinc finger domain protein 1B | BAZ1B | K905↑ | 0.65 |
| AJAP15703 | protein binding and apoptosis | peptidyl-tRNA hydrolase 2, mitochondrial | PTH2 | K128↑ | 0.93 |
| AJAP23458 | protein binding | dynein heavy chain 10, axonemal isoform X3 | DNAH10 | K909↑ | 0.82 |
| AJAP04502 | DNA binding | N-terminal kinase-like protein | NTKL | K29↑ | 0.86 |
| AJAP17355 | transport activity | melanotransferrin | MELTF | K85↑ | 1.40 |
| AJAP15405 | glycolytic process | acyl-CoA dehydrogenase family member 10 | ACAD10 | K416↑ | 0.62 |
| AJAP21096 | threonine catabolic | glycine C-acetyltransferase | GCAT | K97↑ | 1.30 |
| AJAP15667 | oxidation-reduction | NADPH:quinone reductase or related Zn-dependent oxidoreductase | NQR | K56↑ | 0.72 |
| AJAP10667 | xylulose catabolic | xylulokinase | XKS | K359↑ | 1.82 |
| AJAP24097 | DNA integration | Retrovirus-related Pol polyprotein from transposon/ Retro element 1 | RE1 | K30↑ | 1.26 |
| AJAP20997 | tRNA modification | queuine tRNA-ribosyltransferase-like | TGTL | K49↑ | 0.51 |
| AJAP07945 | protein binding | spectrin beta | Spectrinβ | K120↓ | −0.70 |
| AJAP19047 | oxidation-reduction | 2-oxoglutarate dehydrogenase E1 component DHKTD1 | DHKTD1 | K573↓ | −1.21 |
| AJAP05715 | transferase activity | Serine hydroxymethyltransferase | SHMT | K387↓ | −1.21 |
| AJAP19958 | hydrolysis activity | cholinesterase 1 | CHLE1 | K63↓ | −0.41 |
| AJAP17228 | -- | hypothetical protein | -- | K1127↓ | −0.76 |
| Accession No. | Function | Protein Name | Short Name | Acetylated Site | log2value |
|---|---|---|---|---|---|
| AJAP05305 | KAT KADC | CREB-binding protein | CBP | K1583↑ | 0.64 |
| AJAP05305 | KAT KADC | CREB-binding protein | CBP | K1590↑ | 0.64 |
| AJAP05305 | KAT KADC | CREB-binding protein | CBP | K1766↓ | −1.28 |
| AJAP23933 | KAT KADC | chromatin modification-related protein Eaf6 | EAF6 | K60↓ | −0.96 |
| AJAP26676 | protein folding | heat shock protein 90 | HSP90 | K440↑ | 0.96 |
| AJAP22974 | protein folding | T-complex protein 1 | TCP1 | K206↑ | 0.68 |
| AJAP29146 | protein folding | caseinolytic peptidase B protein homolog (HSP78) | CLPB | K576↓ | −1.38 |
| AJAP27049 | protein folding | cyclophilin A (peptidyl-prolyl cis-trans isomerase) | CYP1 | K47↓ | −1.28 |
| AJAP27049 | protein folding | cyclophilin A (peptidyl-prolyl cis-trans isomerase) | CYP1 | K80↓ | −0.69 |
| AJAP04150 | cell protection | glutathione S-transferase | GST | K87↑ | 0.51 |
| AJAP22222 | protein biosynthesis | translation initiation factor 2 subunit 1 | IF2 | K178↑ | 2.04 |
| AJAP06040 | protein biosynthesis | translation elongation factor EF-1alpha (GTPase) | EF1α | K338↓ | −1.12 |
| AJAP15989 | transporter activity | solute carrier family 39 member 10 (zinc transporter ZIP10-like) | SLC39A10 | K43↑ | 1.20 |
| AJAP16427 | transporter activity | sodium-dependent organic anion transporter | NPT | K114↑ | 0.59 |
| AJAP03428 | apoptosis | ELL-associated factor 2-like | EAF2 | K259↑ | 1.13 |
| AJAP01277 | apoptosis | MAP kinase-activating death domain protein-like | MADD | K419↓ | −0.66 |
| AJAP20633 | oxidation-reduction | 2-hydroxyglutarate dehydrogenase | 2HDH | K393↑ | 0.62 |
| AJAP22233 | oxidation-reduction | gamma-butyrobetaine dioxygenase | BBOX | K479↓ | −2.72 |
| AJAP21026 | cytoskeletal binding | lamin B | LMNB | K51↑ | 1.10 |
| AJAP27911 | cytoskeletal binding | tropomyosin | TPM | K365↑ | 1.04 |
| AJAP09828 | cytoskeletal binding | myosin | MYH | K31↓ | −0.63 |
| AJAP21063 | regulation of cytoskeleton | bromodomain and WD repeat-containing protein 3-like, partial | BRWD3 | K81↑ | 1.09 |
| AJAP04260 | DNA integration | transposon Ty3-I Gag-Pol polyprotein | TY3B-I | K272↑ | 0.63 |
| AJAP04502 | retrograde vesicle-mediated transport | N-terminal kinase-like protein | NTKL | K29↑ | 0.97 |
| AJAP20141 | cofactor binding | cytohesin-3-like, partial | CYTH3 | K569↑ | 1.27 |
| AJAP28079 | glycolytic process | fructose-bisphosphate aldolase, class I | FBA1 | K219↑ | 0.65 |
| AJAP00341 | metabolic process | extracellular signal-regulated kinase 2 | ERK2 | K220↑ | 1.24 |
| AJAP12117 | amino acid metabolism | branched-chain-amino-acid aminotransferase-like protein 1 | BCAT1 | K746↑ | 1.08 |
| AJAP17627 | ATP binding | multidrug resistance-associated protein 5 | MRP5 | K51↑ | 0.81 |
| AJAP13674 | ribosome | 60S ribosomal protein L23a | Ribosomal_L23a | K59↓ | −1.12 |
| AJAP09062 | ribosome | large subunit ribosomal protein L3e | Ribosomal_L23e | K225↓ | −1.35 |
| AJAP23675 | ribosome | Ribosomal protein S5 | Ribosomal_S5 | K115↓ | −1.17 |
| AJAP04718 | endoplasmic reticulum membrane | putative helicase MOV-10 | MOV-10 | K349↓ | −0.81 |
| AJAP24952 | transaminase activity | SH2 domain-containing protein 4a-like, partial | SH2D4 | K313↓ | −1.41 |
| AJAP05049 | DNA helicase activity | RuvB-like protein 1 (pontin 52) DNA helicase TIP49, TBP-interacting protein | RUVBL1 | K453↓ | −0.60 |
| AJAP07945 | protein binding | spectrin beta | SPTB | K120↓ | −0.46 |
| AJAP10927 | protein binding | phosphodiesterase family member 7-like | ENPP7 | K840↓ | −1.01 |
| AJAP26499 | nucleic acid binding | RNA recognition motif (RRM) domain | RRM | K101↓ | −0.74 |
| AJAP27984 | nucleic acid binding | R3H domain-containing protein 4 | R3HD4 | K79↓ | −1.00 |
| AJAP07965 | -- | hypothetical protein | -- | K933↓ | −0.56 |
| AJAP24653 | -- | hypothetical protein | -- | K916↓ | −0.88 |
| Accession No. | Function | Protein Name | Short Name | Acetylated Site | log2value |
|---|---|---|---|---|---|
| AJAP22974 | protein folding | T-complex protein 1 | TCP1 | K206↑ | 0.93 |
| AJAP01782 | chromosomal stability | Fanconi anemia group J protein homolog | FANCJ | K274↓ | −0.50 |
| AJAP06040 | protein biosynthesis | translation elongation factor EF-1alpha (GTPase) | EF1α | K192↑ | 0.91 |
| AJAP16427 | transporter activity | sodium-dependent organic anion transporter | NPT | K114↑ | 1.01 |
| AJAP03608 | transporter activity | solute carrier family 25 (mitochondrial adenine nucleotide translocator) | SLC25 | K95↑ | 0.44 |
| AJAP15989 | transporter activity | solute carrier family 39 member 10 (zinc transporter ZIP10-like) | SLC39A10 | K43↑ | 1.06 |
| AJAP17787 | transporter activity | solute carrier family 28 member 3-like | SLC28 | K370↑ | 0.29 |
| AJAP09671 | transporter activity | cyclic nucleotide-gated cation channel alpha-3-like | CNGA3 | K669↑ | 0.97 |
| AJAP09671 | transporter activity | cyclic nucleotide-gated cation channel alpha-3-like | CNGA3 | K671↑ | 0.97 |
| AJAP29476 | transporter activity | zinc finger BED domain-containing protein 4-like | Zbed4 | K117↑ | 0.73 |
| AJAP13059 | transporter activity | zinc finger MYM-type protein 1-like | Zmym1 | K119↓ | −1.10 |
| AJAP17356 | transporter activity | transferrin-like domain | TFD | K42↓ | −2.03 |
| AJAP17356 | transporter activity | transferrin-like domain | TFD | K674↓ | −1.54 |
| AJAP17355 | transporter activity | melanotransferrin | MELTF | K85↓ | −1.60 |
| AJAP18698 | oxidation-reduction | Thiol-disulfide isomerase/thioredoxin | TRX | K51↑ | 0.97 |
| AJAP20633 | oxidation-reduction | 2-hydroxyglutarate dehydrogenase | 2HDH | K393↑ | 0.77 |
| AJAP22233 | oxidation-reduction | gamma-butyrobetaine dioxygenase | BBOX | K479↓ | −2.58 |
| AJAP15456 | autophagy | autophagy-related protein 18 | ATG18 | K203↑ | 0.80 |
| AJAP12353 | apoptosis | ADAMTS-like protein 2 isoform X1 | Adamtsl2 | K621↓ | −0.41 |
| AJAP01277 | apoptosis | MAP kinase-activating death domain protein-like | MADD | K419↓ | −0.87 |
| AJAP28079 | protein binding | fructose-bisphosphate aldolase, class I | ALDOA | K60↑ | 0.95 |
| AJAP11248 | steroid metabolic | steroid 17-alpha-hydroxylase/17,20 lyase | CYP17 | K75↑ | 1.24 |
| AJAP09155 | cholesterol biosynthetic | 3-keto-steroid reductase/17-beta-hydroxysteroid dehydrogenase 7 | HSD17b7 | K200↑ | 0.92 |
| AJAP19958 | hydrolysis activity | cholinesterase 1 | CHLE1 | K63↑ | 0.60 |
| AJAP19958 | hydrolysis activity | cholinesterase 1 | CHLE1 | K191↓ | −0.97 |
| AJAP27911 | cytoskeletal protein binding | tropomyosin | TPM | K365↑ | 1.07 |
| AJAP14831 | hydrolase activity | acyl-coenzyme A thioesterase 9 | ACOT9 | K93↑ | 1.11 |
| AJAP15858 | ribosome | 39S ribosomal protein L9, mitochondrial isoform X3 | Ribosomal_L9 | K191↑ | 1.17 |
| AJAP17052 | ribosome | large subunit ribosomal protein L10e | Ribosomal_L10e | K101↓ | −0.76 |
| AJAP05814 | ribosome | ribosomal protein S12/S23 | Ribosomal_S12/S23 | K44↓ | −0.62 |
| AJAP15405 | glycolytic process | acyl-CoA dehydrogenase family member 10 | ACAD10 | K416↓ | −0.67 |
| AJAP21933 | glycolytic process | pyruvate kinase | PKA | K100↓ | −0.62 |
| AJAP29031 | negative regulation of translation | GRB10-interacting GYF protein 2 | GIGYF2 | K147↓ | −1.29 |
| AJAP15703 | protein binding | peptidyl-tRNA hydrolase 2, mitochondrial | PTH2 | K128↓ | −1.48 |
| AJAP07321 | metal binding | serum paraoxonase/arylesterase 1-like | PON | K201↓ | −1.62 |
| AJAP26236 | metal binding | pol-like protein | POLL | K338↓ | −0.57 |
| AJAP08618 | GTPase activation | Rab GDP dissociation inhibitor | GDI | K147↓ | −0.55 |
| AJAP09462 | G-protein coupled receptor | alpha-1A adrenergic receptor | ADA1A | K123↓ | −1.10 |
| AJAP16917 | -- | hypothetical protein | -- | K345↓ | −1.59 |
| AJAP16076 | -- | hypothetical protein | -- | K131↑ | 1.19 |
| AJAP05930 | -- | hypothetical protein | -- | K174↑ | 1.44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Wang, X. Lysine Acetylation is an Important Post-Translational Modification that Modulates Heat Shock Response in the Sea Cucumber Apostichopus japonicus. Int. J. Mol. Sci. 2019, 20, 4423. https://doi.org/10.3390/ijms20184423
Xu D, Wang X. Lysine Acetylation is an Important Post-Translational Modification that Modulates Heat Shock Response in the Sea Cucumber Apostichopus japonicus. International Journal of Molecular Sciences. 2019; 20(18):4423. https://doi.org/10.3390/ijms20184423
Chicago/Turabian StyleXu, Dongxue, and Xuan Wang. 2019. "Lysine Acetylation is an Important Post-Translational Modification that Modulates Heat Shock Response in the Sea Cucumber Apostichopus japonicus" International Journal of Molecular Sciences 20, no. 18: 4423. https://doi.org/10.3390/ijms20184423
APA StyleXu, D., & Wang, X. (2019). Lysine Acetylation is an Important Post-Translational Modification that Modulates Heat Shock Response in the Sea Cucumber Apostichopus japonicus. International Journal of Molecular Sciences, 20(18), 4423. https://doi.org/10.3390/ijms20184423