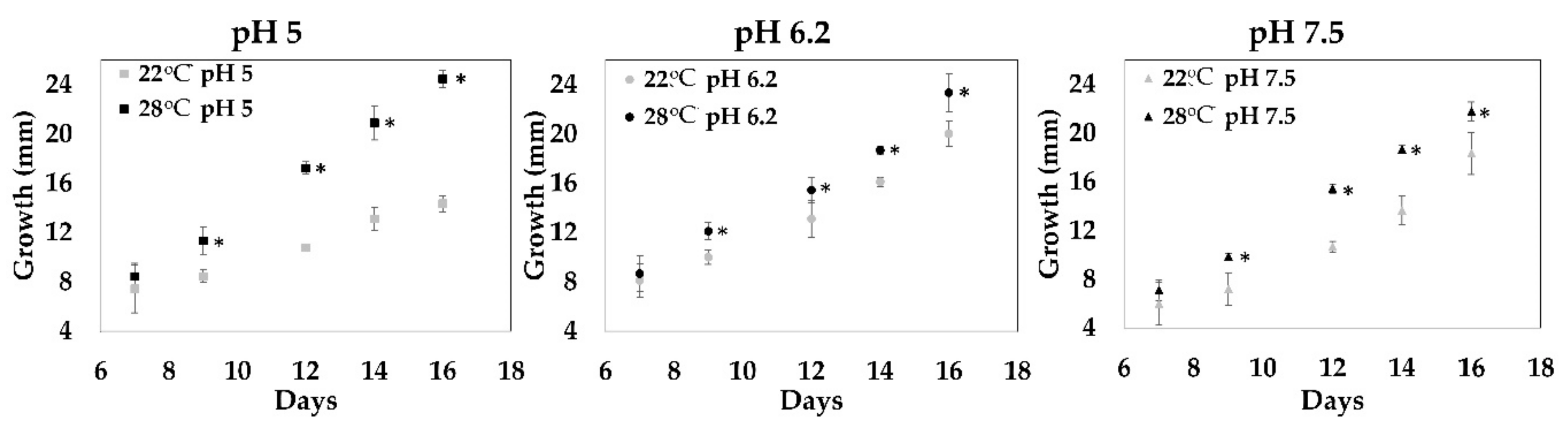
2.1. Optimal Growth Conditions
The sclerotia of
I. obliquus grow very slowly in nature, and artificial culture is difficult. To produce large amounts of biomass,
I. obliquus was cultured using different growth conditions. Mycelial growth tests were carried out on a culture medium to determine optimal conditions. The growth of the
I. obliquus mycelium was conducted under nine conditions (three different pH (5, 6.2 and 7.5) and three different temperatures (22, 28, and 37 °C) over 16 days in a solid medium.
Figure 1 shows the results for two temperatures because at 37 °C, no growth of
I. obliquus mycelium was observed (data not shown). At 28 °C,
I. obliquus growth was significantly higher for all tested pH; however, pHs 5 and 6.2 showed the best results (
Figure 1). The effect of temperature on chaga’s growth was only observed at pH 5. For example, on day 16 at pH 5 in the solid culture, fungal growth at 28 °C was twice the growth observed at 22 °C (
Figure 1; from 12 mm at 22 °C to more than 24 mm at 28 °C). However, the effect of pHs 5 or 6.2 on fungal growth at 28 °C was not significantly different (
Figure 1; circa 24 mm for both pH).
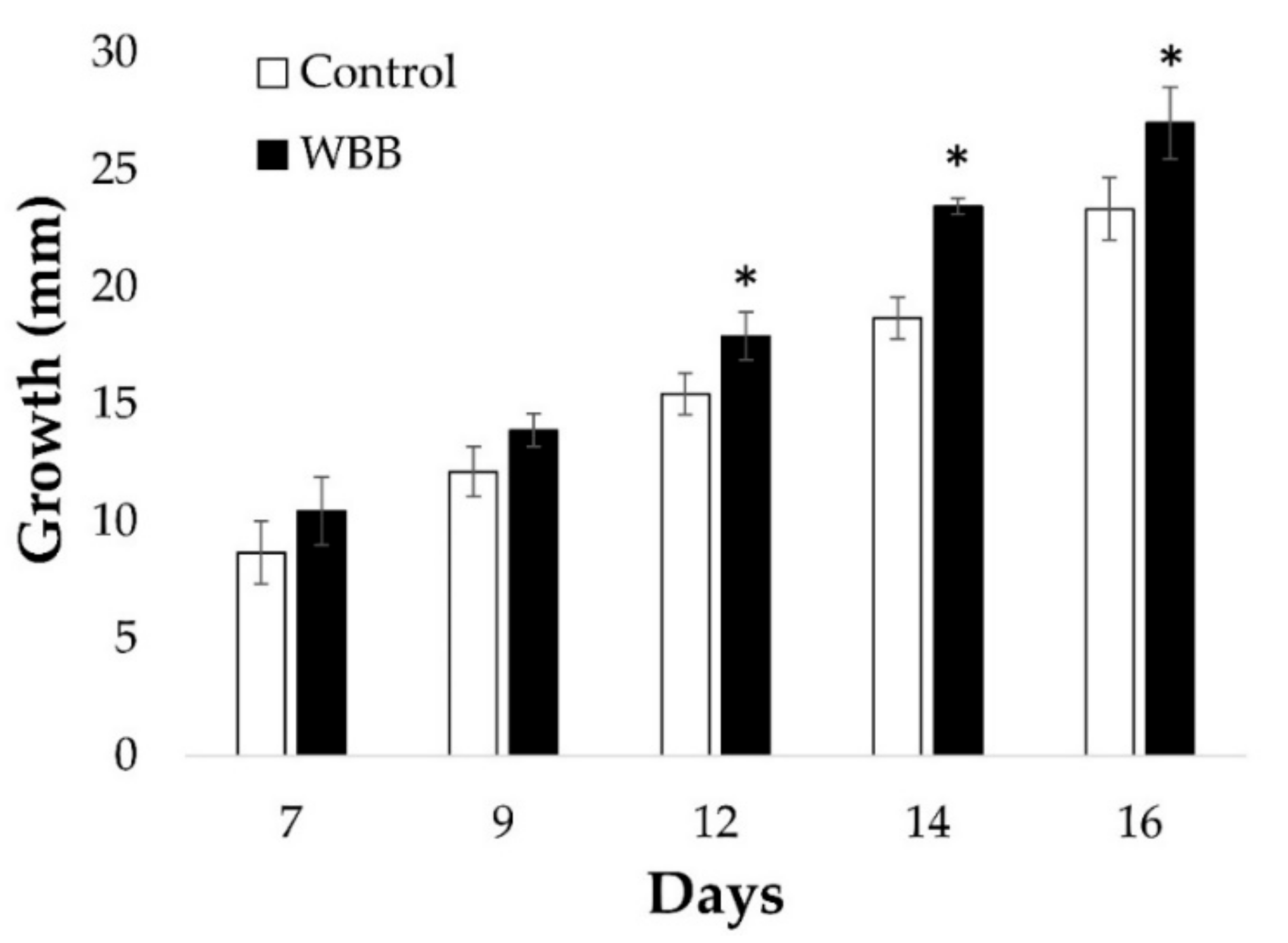
In the wild, the growth and production of terpenoids by
I. obliquus is likely influenced by substrates provided by its host. Thus, an investigation to determine the influence of white birch bark (WBB) on growth of
I. obliquus under optimal pH and temperature conditions was carried out. For this purpose,
I. obliquus cells were grown in presence or absence of WBB residues. Results showed that
I. obliquus growth was significantly higher in presence of WBB (
Figure 2). Specifically, after 16 days of co-culture, the
I. obliquus control mycelia reached 23 mm diameter, whereas those that were WBB-treated amounted to 27 mm (
Figure 2). This indicates that
I. obliquus growth was stimulated by the presence of bark residues from its host.
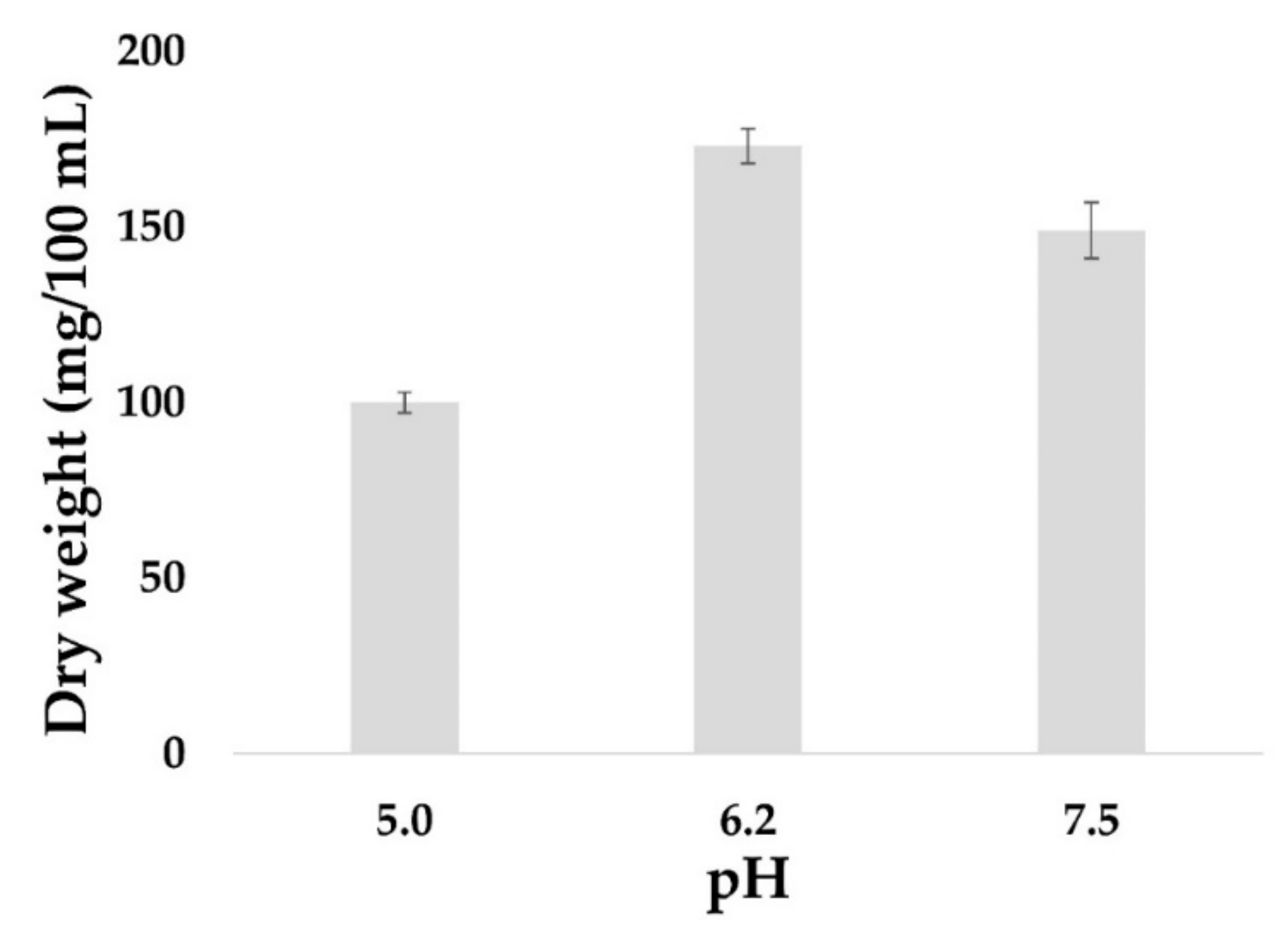
The yield of fungal biomass is often higher in a liquid culture compared to a solid culture. Furthermore, a liquid culture is easier to manipulate in order to extract and obtain cell components such as metabolites, proteins or nucleic acids. Thus, the effect of pH on the kinetics of mycelial growth of
I. obliquus in a liquid medium at 28 °C was investigated. A rapid increase in the biomass was detected within the first six days, concurrent with the cells being in the exponential growth phase with significant consumption of carbon. On day eight, the mycelial biomass reached the apex, and the optimal growth of
I. obliquus was observed at pH 6.2 with 0.18 g/100 mL of mycelial dry weight compared to 0.10 g/100 mL at pH 5 and 0.14 g/100 mL at pH 7.5 (
Figure 3).
By-products of metabolism are shuttled throughout the cell and utilized to grow. Thus, optimal growth may influence the production of terpenoids [
46]. In this study, different culturing conditions were used to establish the best possible parameters for
I. obliquus growth. According to the results, the best condition for
I. obliquus growth is at pH 6.2 and at 28 °C in a liquid culture that continuously shaken at 150 rpm. These results are supported by other studies on
I. obliquus reporting similar optimal growth conditions [
46,
50,
51]. The results of growth in the liquid and solid culture medium support that pH 6.2 would be the optimal pH for optimal mycelium growth.
For subsequent analyses, an I. obliquus liquid culture grown at 28 °C and pH 6.2 were used, since they showed optimal growth and morphological properties. In addition, it provided an easier system for the collection of cells for RNA extraction.
2.2. Illumina Sequencing and de Novo Assembly
To better understand the molecular mechanisms underlying the differences in chaga’s transcriptome caused by the presence of terpenoid substrates, RNA-Seq was performed. RNA was extracted from three biological replicates of cultured
I. obliquus in three different conditions: In the absence (control,
n = 3) or presence of the terpenoid substrate betulin (BET,
n = 3) or white birch bark (WBB,
n = 4). Ten corresponding cDNA libraries were generated and sequenced using Illumina HiSeq 4000 PE100. The raw reads for each library were deposited to the NCBI Sequence Read Archive under the accession PRJNA526077 (
Appendix A—
Table A1). A total of 229,970,255 raw paired reads were generated from all replicates of the three conditions of culturing. After filtering out low-quality sequences, 219,288,500 clean reads were obtained, corresponding to approximately 95% of the total raw reads (
Appendix A—
Table A1). Because of the lack of availability of information (i.e., genome, transcriptome) on
I. obliquus or its related species, we combined all RNA-Seq libraries to build a deep transcriptome using de novo assembly. All clean reads obtained from the ten libraries were subsequently de novo assembled using the Trinity program (version 2.6.5), and a total of 196,273 transcripts with an average length of 2521 bp, and an N50 length of 4052 bp were obtained (
Appendix A—
Table A1). An evaluation of the size distribution showed that 96% of all transcripts of
I. obliquus have lengths longer than 1 kb (
Appendix A—
Figure A2).
In a previous study, Zou et al. (2016) reported on the Illumina sequencing of
Inonotus baumii [
52] and obtained a total of 27,259,264 reads. After the Trinity de novo assembly, 30,051 unigenes with an average length of 561 bp and an N50 length of 831 bp were generated, which is two-to-three times less than our results [
52]. In another research work on the saprophytic fungus
Wolfiporia cocos, the Illumina sequencing of the transcriptome yielded a total of 38,722,186 reads, which were assembled into 60,354 contigs with an N50 of 765 bp [
53]. In comparison to this, we obtained eight-to-ten times more reads, ensuring more coverage and allowing for a more contiguous assembly, as confirmed by the average transcript length of 2521 bp obtained here (
Appendix A—
Table A1).
The average transcript lengths of eukaryotic genes were greater than 1 kb. For example, the average transcript lengths of eukaryotic genes range from 1108 to 2667 bp in human and from 1135 to 1695 bp in yeast [
54]. Additionally, eukaryotic proteins have an average size of 472 amino acid residues (i.e., 1419 bp), although the size of proteins from plant genomes are smaller than those of fungi and animals [
55]. Based on our sequencing results, it could be concluded that the quality and the depth of this transcriptomic assembly is significantly improved compared to other fungal transcriptome studies.
2.3. Functional Annotation of the RNA-Seq Data
After the Basic Local Alignment Serach Tool (BLAST) annotation of the de novo assembly against the uniprot_sprot.trinotate_v2.0.pep protein database, a total of 20,072 transcripts with an average length of 4502 bp and an N50 length of 5512 bp were obtained (
Appendix A—
Table A1). A large number of transcripts had a similarity with known genes, suggesting a large amount of sequences specific to
I. obliquus.
Gene function was annotated based on the following databases: Gene Ontology (GO), Clusters of Orthologous Groups of proteins (COG), and Protein family database (Pfam). A total of 86,246 transcripts (43.94% of assembled transcripts) had a match in the GO database with an E-value of 0 (
Table 1). Additionally, 52,224 transcripts (20.05% of assembled transcripts) and 38,415 transcripts (19.57%) showed similarity to sequences in the COG and Pfam databases, respectively (
Table 1).
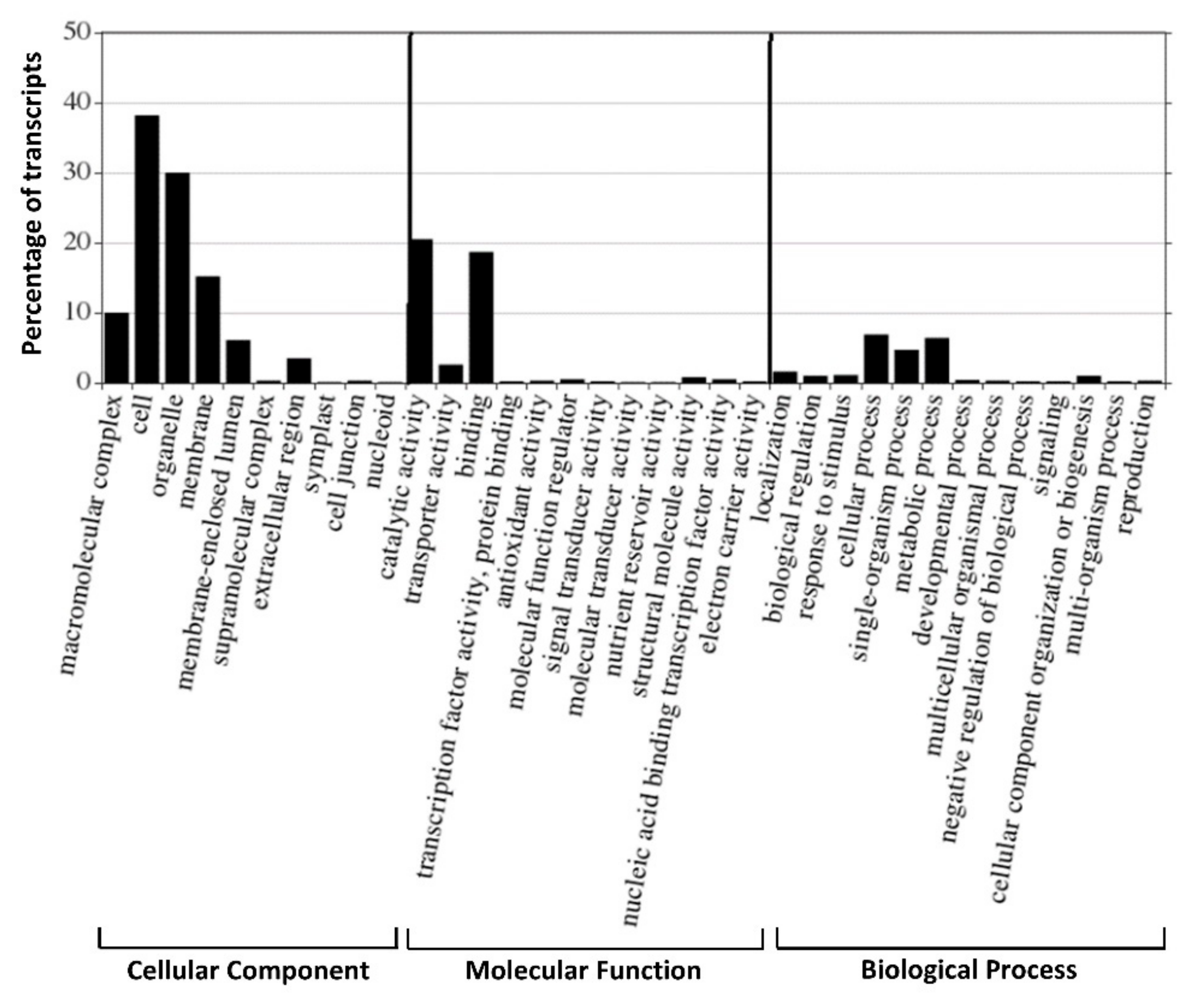
The functional GO classification of genes is considered to be an effective tool, offering controlled vocabulary and strictly defined biological process for annotating and performing functional analysis of a large number of annotated genes and their products in a selected organism [
56]. The GO classification was performed on the 196,273 transcripts, and a total of 86,246 transcripts were classified into 41 functional groups, categorized into three main GO ontologies: Biological process, cellular component and molecular function. The results indicated that the most highly annotated GO category was the cellular component (49.63%) with cell, organelle and membrane being the most abundant, while only a few transcripts were attributed to symplast, cell junction and nucleoid (
Figure 4). Under the category of molecular function (38.42%), catalytic activity and binding activity were, respectively, the largest categories. Regarding biological process (11.95%), the dominant subcategories included genes involved metabolic process and cellular process (
Figure 4). In addition, important categories with low numbers of transcripts were also represented in the GO, such as transcription factor activity, protein binding and nucleic acid binding.
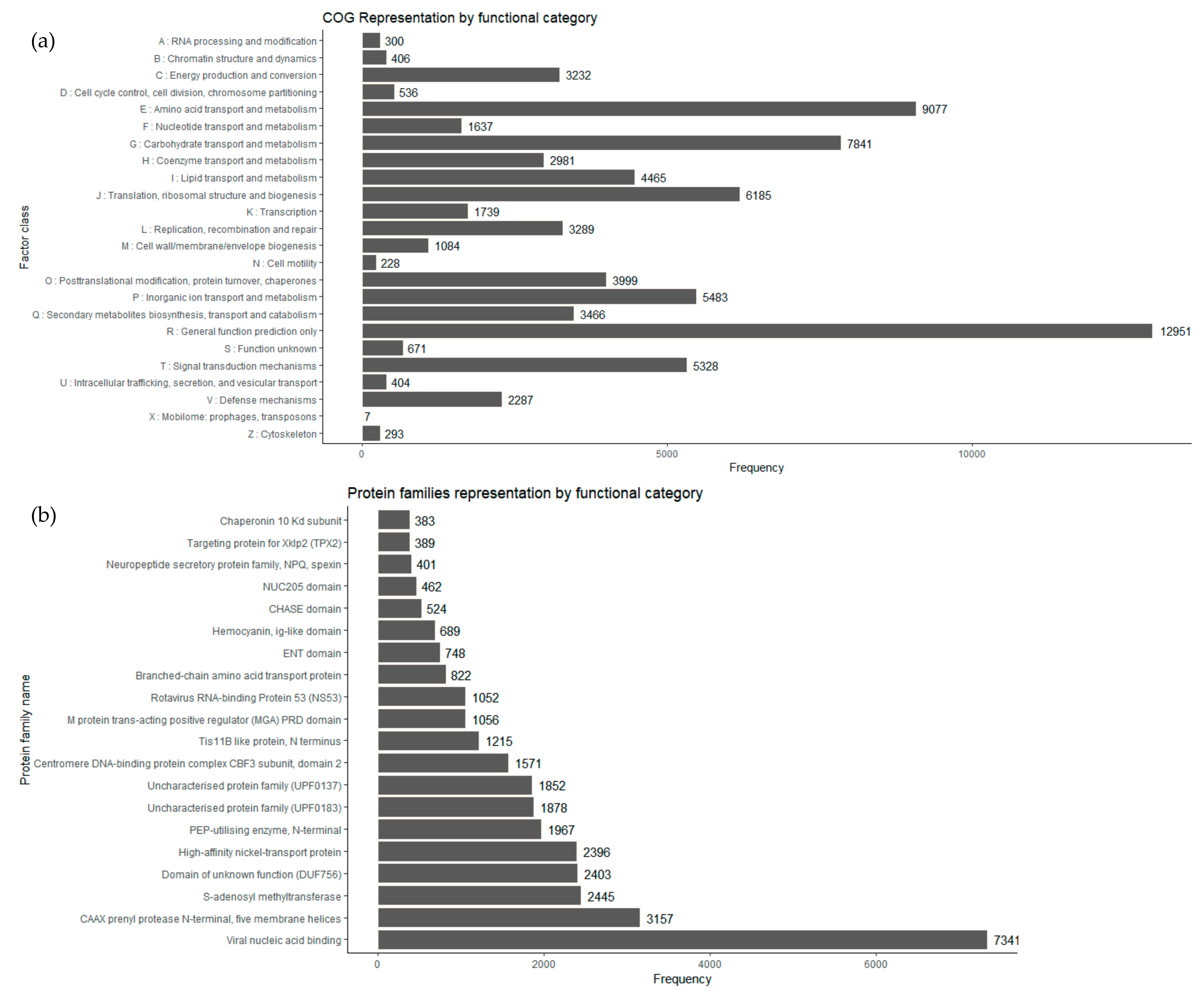
COG is a database where gene products from a common family ancestor are classified. From the
I. obliquus transcriptome, COG-annotated putative proteins were functionally classified into, at least, 25 protein families involved in basic function, such as transcription, translation, signal transduction, cellular structure, biochemistry, metabolism, and molecular processing. The COG analysis of
I. obliquus transcriptome led to the classification of 52,224 transcripts to COG classifications. The top COG categories included basic function prediction (16.63%), followed by amino acid transport and metabolism (11.65%), carbohydrate transport and metabolism (10.06%), translation, ribosomal structure and biogenesis (7.94%), inorganic ion transport and metabolism (7.03%), signal transduction, mechanisms (6.84%) and posttranslational modification, chaperones (5.13%) (
Figure 5a). It is interesting that 4.45% of the
I. obliquus annotated transcripts were ranked in the specialized metabolites biosynthesis, transport and catabolism, which suggests that specialized metabolism occurs in cultivated
I. obliquus cells.
Protein domain analyses were performed using the Pfam database [
57]. A total of 38,415 transcripts of
I. obliquus were found to be associated with protein domains (
Figure 5b). Compared to the
I. baumi transcriptome, in which a total of 8276 transcripts contain at least one Pfam protein domain [
52], our
I. obliquus Pfam analysis was six times superior. This result provides another indication that our transcriptome was of good quality and had more depth.
Among the abundant protein families expressed, several were linked to primary metabolism enzymatic activities such as prenylprotease,
S-adenosyl methyltransferase and phosphotransferase (PEP-utilizing enzyme). In addition, an important number of the expressed genes encoded predicted proteins from families implicated in ion transport, more specifically Nickel transport protein, a protein family proven to be essential for energy and nitrogen metabolism [
58].
2.4. Differential Expression Analysis
Differential expression analyses were performed between databases of
I. obliquus. Transcripts with adjusted
p-values ≤0.05 and a fold change (log2FC) ≥1 were designated as significantly differentially expressed transcripts. For
I. obliquus cells cultivated with betulin (control vs. BET), the results showed that 441 differentially expressed transcripts (139 up-regulated and 302 down-regulated) were identified. Twenty-two times more transcripts (9707) were differently expressed in cells cultivated with bark (control vs. WBB), with a total of 5070 annotated transcripts up-regulated and 4637 transcripts down-regulated (
Figure 6). This suggest that the birch bark has very different molecular impacts and requires more transcriptome adjustment from
I. obliquus than betulin.
The top twenty-five up- and down-regulated expressed transcripts in
I. obliquus cells cultivated with betulin (
Appendix A—
Table A2 and
Table A3) or white birch bark (
Appendix A—
Table A4 and
Table A5) were identified. After blasting the top twenty-five up- and down-regulated transcripts against the NCBI database, 48% could not be annotated (no hit). Among top-regulated transcripts with predicted function, 16% have an E-value of 10
−5 or less (
Appendix A—
Table A2,
Table A3,
Table A4 and
Table A5). Interestingly, the transcript annotated as phosphatidylinositol 4-phosphate 3-kinase ranked first up-regulated for both treatments, suggesting the importance of the signaling pathways involved in cell proliferation, cell survival, and intracellular protein trafficking (
Appendix A—
Table A2 and
Table A4). Phosphatidylinositol 4-phosphate 3-kinase is a conserved enzyme involved in the regulation of phosphatidylinositol 4-phosphate, which is crucial for maintaining morphology, regulating lipid storage, Golgi function and actin cytoskeleton organization [
59]. The signaling cascade activated downstream of this enzyme implicates the mitogen-activated protein kinase (MAPK) pathway, which regulates the filamentous growth of yeast [
60]. This suggests a putative role for phosphatidylinositol 4-phosphate 3-kinase in the regulation of the morphology of the mycelium and filamentous growth in
I. obliquus cells in presence of WBB or betulin.
In addition, caffeic acid 3-O methyltransferase, which catalyzes the conversion of caffeic acid to ferulic acid, was up-regulated in both treatments (
Appendix A—
Table A2 and
Table A4). Phenolic acids, such as caffeic acid and ferulic acid, possess antioxidant properties that play key roles in the synthesis of polyphenols and protection against UV and oxidative stress [
6,
61,
62,
63]. The up-regulated condition of caffeic acid 3-
O-methyltransferase in our transcriptomic results suggests an important role for ferulic acid in cultivated
I. obliquus cells for protection or polyphenol synthesis.
The most down-regulated transcripts found in the betulin-cultivated cells were annotated as transcripts involved in cellular processes such as DNA replication, transcription, cell division and proliferation, i.e., t-RNA ligase, cyclin-dependent kinase, ribonucleoprotein complex subunits, chromosome proteins, and condensin complex subunits. This suggests betulin-induced cellular processes in
I. obliquus cells (
Appendix A—
Table A3).
For WBB-cultivated cells (
Appendix A—
Table A5), most down-regulated transcripts found annotated for protein involved in metabolic processes such as several peroxidases, lipases, and racemases. This suggests that cultivation with WBB reduced the expression of genes encoding proteins involved in ligninolysis, i.e., to metabolize substrates/degrade bark molecules [
64]. Effective lignin and plant cell wall degradation is possible through the action of enzymes from filamentous fungi, and it has been reported that fungi of the basidiomycetes family express several enzymes of the peroxidase family in their transcriptomes, such as lignin- and manganese-peroxidases known for their ability to degrade plant cell walls [
65]. The down-regulated peroxidase transcripts in
I. obliquus cells cultivated with WBB suggests that WBB may leak repressors for peroxidases to prevent its degradation from the mycelium.
2.5. Genes Involved in the Biosynthesis of Terpenoids in Cultured I. Obliquus Cells
The presence and identification of terpenoids in
I. obliquus cells have been already studied and are well documented [
1,
22,
28,
35,
40,
41,
43,
46,
66,
67]. In this study, narrow BLAST searches were achieved to identify distinct transcripts encoding enzymes presumably taking part in terpenoid biosynthesis (
Appendix A—
Figure A1). Eighteen transcript sequences from the mevalonate pathway involved in terpenoid biosynthesis were found in the transcriptomic data of
I. obliquus (
Table 2). As expected, no genes involved in the non-mevalonate pathways were identified, suggesting the absence of this pathway in chaga. Similarly, the transcriptome from
I. baumii reported no genes from the non-mevalonate pathway [
52].
From the precursor pathway leading to IPP/DMAPP (
Appendix A—
Figure A1), several transcript variants of orthologous genes were identified (
Table 2). For example,
AACT genes have been cloned and characterized from various species including zebra fish, frog and human. Two human (
Homo sapiens (Hs)) isoforms (HsAACT1 and HsAACT2) were identified to be ubiquitous and important enzymes found in different intracellular locations. The cytosolic HsAACT1 catalyzes the formation of the acetoacetyl-CoA required for sterol, including cholesterol biosynthesis. The exact function of HsAACT2 is not known, but patients with HsAACT2 deficiency have shown severe mental retardation and hypotonus [
68]. Similarly, the model plant
Arabidopsis thaliana (At) has two isoforms of AACT, where
AtAACT1 is primarily expressed in the vascular system and
AtAACT2 is deeply present in root tips, top stems, young leaves, and anthers. The characterization of T-DNA insertion of mutated alleles for each
AtAACT locus established that
AtAACT2 function is necessary for normal male gamete transmission and embryogenesis, whereas plants lacking
AtAACT1 are viable with no apparent growth phenotype [
69]. In yeast,
AACT (also called
erg10) encodes a multimeric enzyme, but the exact subunit structure has not been defined [
68].
Transcripts encoding each enzyme involved in the formation of terpenoid precursors IPP and DMAPP (
Appendix A—
Figure A1) were identified in the transcriptome of
I. obliquus (
Table 2). According to the number of reads, the
HMGS, HMGR, and
PDM transcripts were the most abundant with E values of 0, suggesting that these gene sequences are well conserved.
Sesquiterpene synthases play an essential role in diversifying the skeletal structure of sesquiterpenoids by catalyzing the very complex cyclisation of the common precursor, FPP [
38,
70,
71,
72]. Sesquiterpenoids have been reported in Inonotus [
43]; however, no biosynthetic genes have been identified. BLASTx searches of
I. obliquus transcriptome led to the identification of three sesquiterpene synthases: Two transcript variants of the muurolene synthase (
MUS1 and
MUS2) and one of the protoilludene synthase (
PRS) (
Table 2).
MUS1 and
MUS2 are only 36% identical at the nucleotide level, suggesting different functions, i.e., substrates for enzymatic activities. Similarly, the E values of
MUS1,
MUS2 and particularly
PRS suggest that their function may be different than orthologous sequences, suggesting that
I. obliquus possess the machinery to produce structurally different sesquiterpenoids. Muurolene, protoilludene, and derivatives, also considered volatile organic compounds (VOCs), are produced by several fungal species. The rich bouquet of several VOCs contribute to the aroma and flavor of mushrooms. Though the ecological function of fungal volatiles is unknown, studies showed their biological activities as antibiotics, antioxidants or inhibitor of germination [
72,
73,
74].
Lee et al. (2016) performed a transcriptomic analysis of the white rot fungus
Polyporus brumalis which led to the identification of two transcripts, germacrene A synthase and trichodiene synthase, involved in sesquiterpenoid biosynthesis [
75]. However, the final products of these enzymes were not detected in the cultivated fungus [
75]. To our knowledge, this study is the first to report the presence of transcripts encoding sesquiterpenoid-forming enzymes in Inonotus.
Triterpenoids from Inonotus species include high pharmaceutical valued metabolites. For example, the lanosterol-derived triterpenoid inotodiol and the lupeol derivative betulinic acid are well known bioactive triterpenoids from
I. obliquus [
17,
19,
24,
25,
27,
28,
33,
36,
76,
77]. Though several studies have shown the presence of triterpenoids in
I. obliquus, only one gene, SQS, has been identified [
78]. In order to increase knowledge on genes involved in triterpenoids production, a search for these genes was carried out in the transcriptome of
I. obliquus. Two transcript variants of squalene synthase (
SQS1 and
SQS2) were identified with a 95% sequence identity to each other (
SQS2 has an insertion at the 3′ end) (
Table 2). Our
SQS1 transcript sequence is identical to previously characterized
SQS from
I. obliquus [
78].
A single transcript for
AO was identified in the transcriptome of
I. obliquus, whereas two transcript variants,
LAS1 and
LAS2, were found (
Table 2).
LAS1 and
LAS2 possess a 98% sequence identity, but
LAS2 is longer and far less expressed than
LAS1, suggesting the importance of
LAS1 in the production of lanosterol and its derivatives in
I. obliquus cell cultures (
Table 2).
According to the literature, only one terpenoid gene,
SQS, has been characterized from
I. obliquus, whereas nine genes related to terpenoids biosynthesis where identified in
I. baumii [
52,
78]. The transcriptome generated in the current study allowed for the identification of all (17) orthologous genes involved in terpenoid metabolism. Furthermore, variants of the
AACT,
MUS,
SQS, and
LAS genes were identified, indicating that these steps may be regulated differently in
I. obliquus. For example, the higher expression of
AACT1,
MUS1, and
LAS1 suggests the importance of theses isoforms in terpenoid formation of
I. obliquus-cultivated cells.
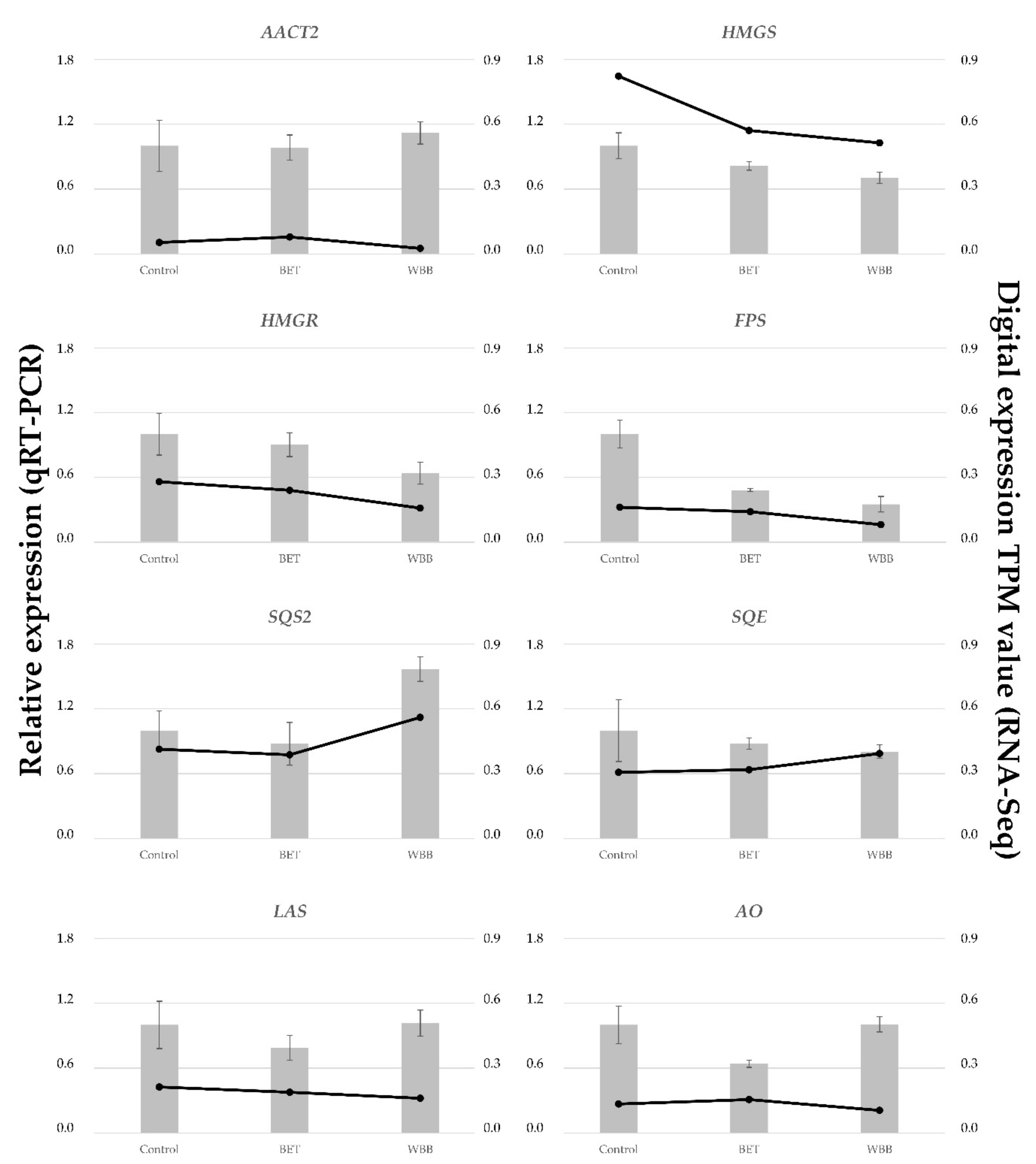
The digital expression of each terpenoid-associated transcript was studied for each growth condition (
Figure 7). Upstream precursor genes, such as
AACT1 and
PMD, had a higher expression in
I. obliquus cells compared to specific downstream (sesqui and triterpenoid) genes such as
PRS and
LAS2 (
Figure 8). From the precursor pathway, the most strongly expressed transcript,
AACT1, remained highly expressed across the different treatments, suggesting no transcriptional regulation under those conditions, whereas
PMD,
SQS2 and
SQE were up-regulated in presence of BET and WBB (
Figure 7). In contrast,
HMGR,
MUS1, and
AO were down-regulated in presence of the terpenoid substrate, particularly in
I. obliquus cells cultivated with WBB. HMGR is a rate-limiting enzyme in the terpenoid biosynthesis. It is possible that the addition of terpenoid substrates promotes negative feedback regulation mechanisms. For example, HMGR, which generates mevalonate, a critical intermediate in the biosynthesis of terpenoids, is exposed to large amount of feedback regulation through various mechanisms that are influenced by end-products, light, and hormones [
79,
80,
81,
82].
2.6. qRT-PCR Validation of RNA-Seq Gene Expression Data
To validate the RNA-Seq digital expression data, eight transcripts encoding terpenoid biosynthetic enzymes were selected for the qRT-PCR analysis. For control reference genes, transcripts with no/low variation were extracted from the database using a custom method developed by dos Santos et al. (2019), which has shown to outperform pre-defined references genes [
83]. Indeed,
centromere protein 3 (
CEN3) and
glutamine RNA ligase (
GluRL) show no/low difference of expression among the conditions tested indicating that they are good reference genes for qRT-PCR study (
Figure 7).
All eight transcripts qRT-PCR analyses are consistent with RNA-Seq results (
Figure 8). This indicates that our transcriptome was reliable and that we could make reasonable inferences from the differentially expressed transcripts. For example, as observed for the digital expression,
SQS2 was up-regulated and
HMGR was down-regulated in presence of terpenoid substrates (
Figure 7 and
Figure 8).
Interestingly, qRT-PCR analysis showed a downregulation of FPS in the presence of terpenoid substrates, suggesting a negative feedback regulation on this important step in the sesqui and triterpenoid biosynthesis. In contrast, SQS2 expression increased in the presence of WBB, suggesting a positive regulation. This opposite regulation, negative on FPS and positive on SQS2, may be due to the presence of terpenoids such as FPP in bark residues. Altogether, the results suggest that few key reactions in the terpenoid pathway are regulated at the transcriptional level in presence of terpenoid substrates in I. obliquus cell cultures.
The amounts of valuable terpenoids accumulated remained low under exogenous induction in a fungal culture. The metabolic engineering of microorganisms is an interesting alternate route for the production of these important compounds. To do this, a better understanding of the terpenoid biosynthetic pathway and the genes involved in this pathway is required. However, the lack of genomic information about I. obliquus hinders the development of alternative production methods. In this work, the best conditions for growth of I. obliquus cells cultivated in presence of terpenoid substrates, betulin or white birch bark were examined, and the corresponding transcriptomes obtained after next-generation sequencing and de novo assembly. Transcriptome analysis identified eighteen transcripts encoding enzymes implicated in the terpenoid metabolism. Comparative analyses of the transcriptomes yielded valuable information with respect to the wide variety of genes implicated in terpenoid metabolism in I. obliquus cells under the described conditions. It would be interesting to compare the laboratory-adapted isolate of I. obliquus to the wild type fungus to monitor gene expression implicated in the host-pathogen interaction between chaga and its host, birch trees.

 and
and 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








