MET Receptor Tyrosine Kinase Regulates the Expression of Co-Stimulatory and Co-Inhibitory Molecules in Tumor Cells and Contributes to PD-L1-Mediated Suppression of Immune Cell Function


 ,
,
Abstract
:1. Introduction
2. Results
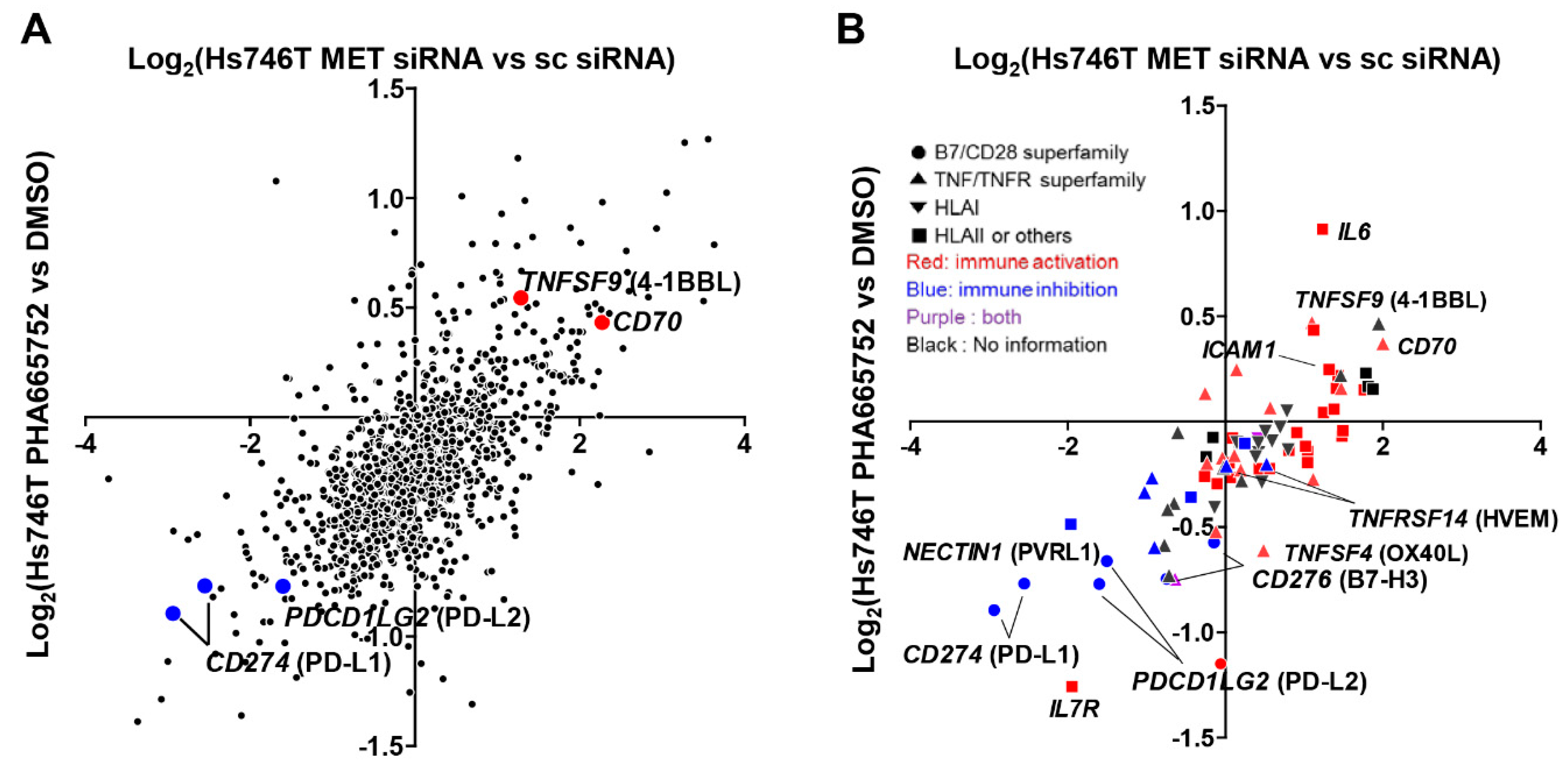
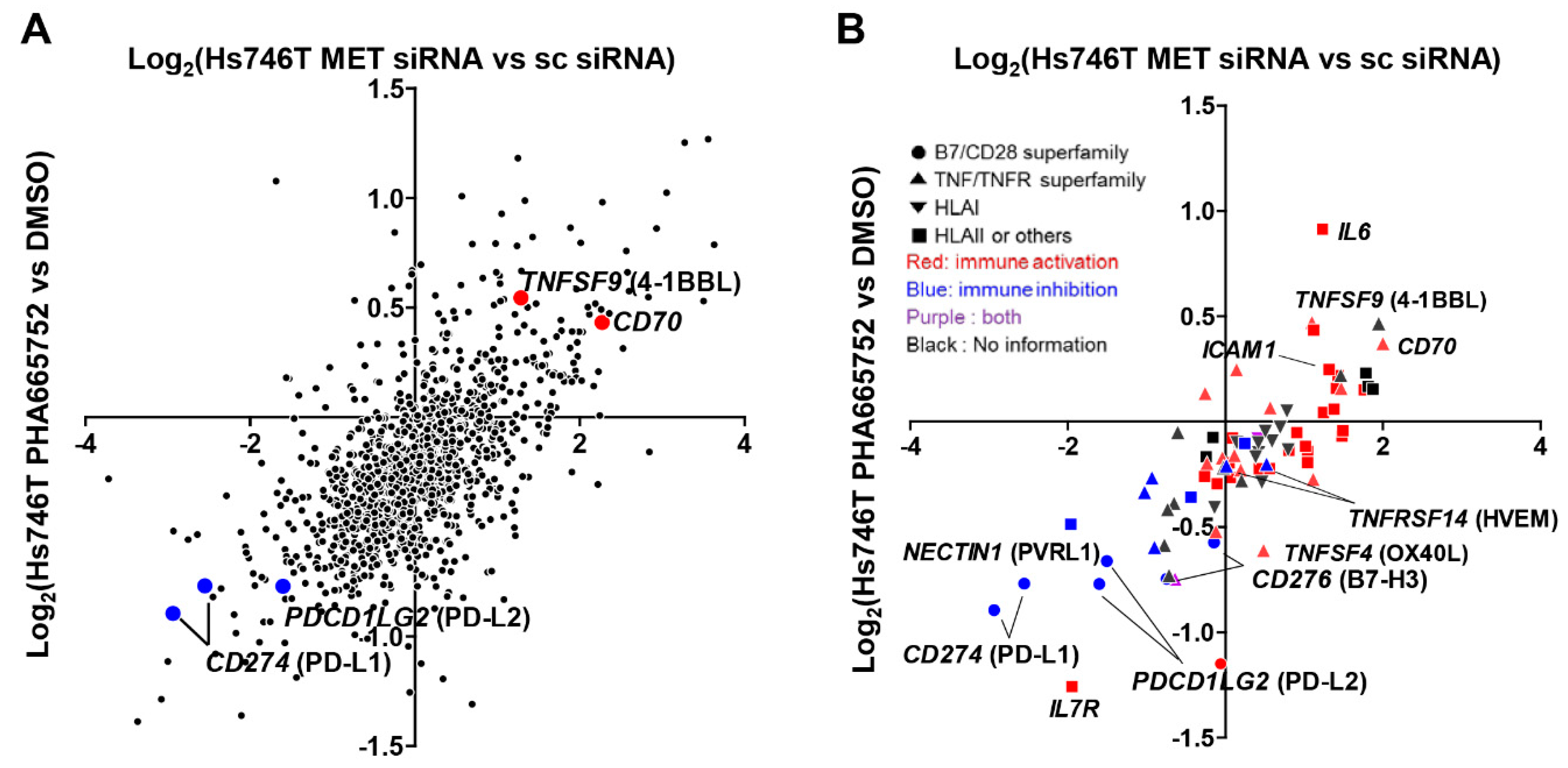
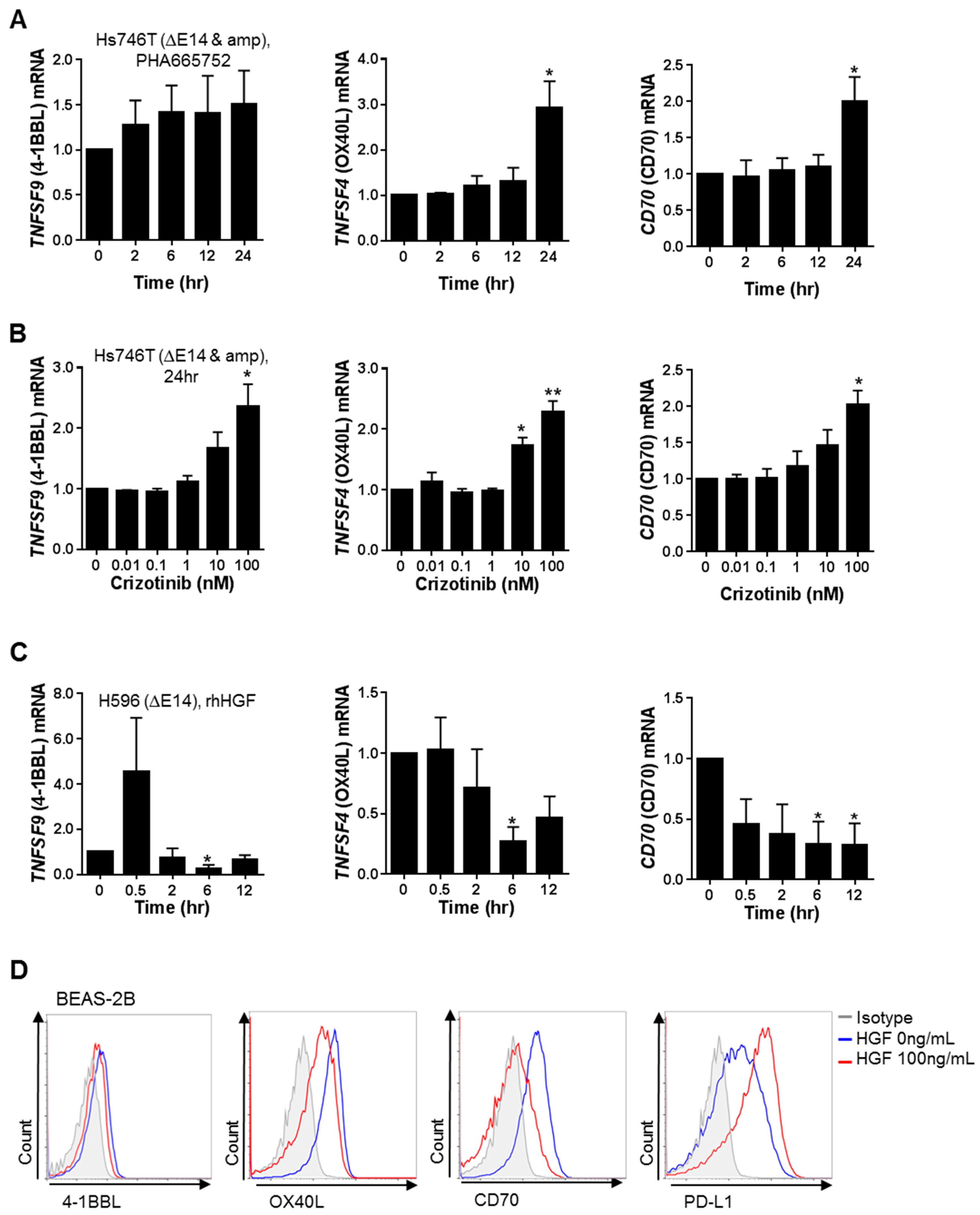
2.1. MET Inhibition or Knockdown in Hs746T Cells Causes Changes in the Expression of Immune-Response-Related Genes
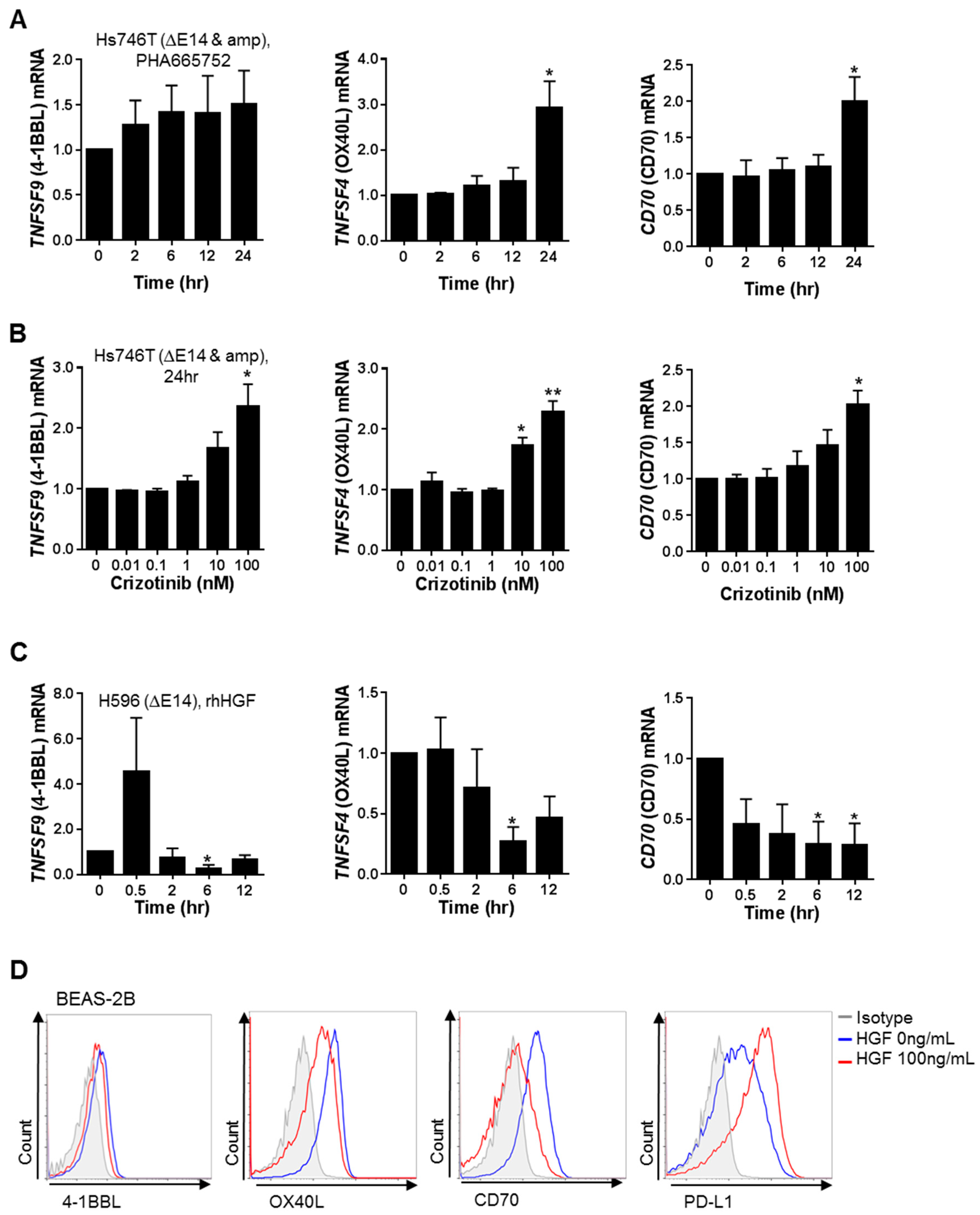
2.2. MET Down-Regulates the Expression of Co-Stimulatory Molecules and Up-Regulates the Expression of Co-Inhibitory Molecules, Particularly PD-L1, in Tumor and Non-Neoplastic Bronchial Epithelial Cells
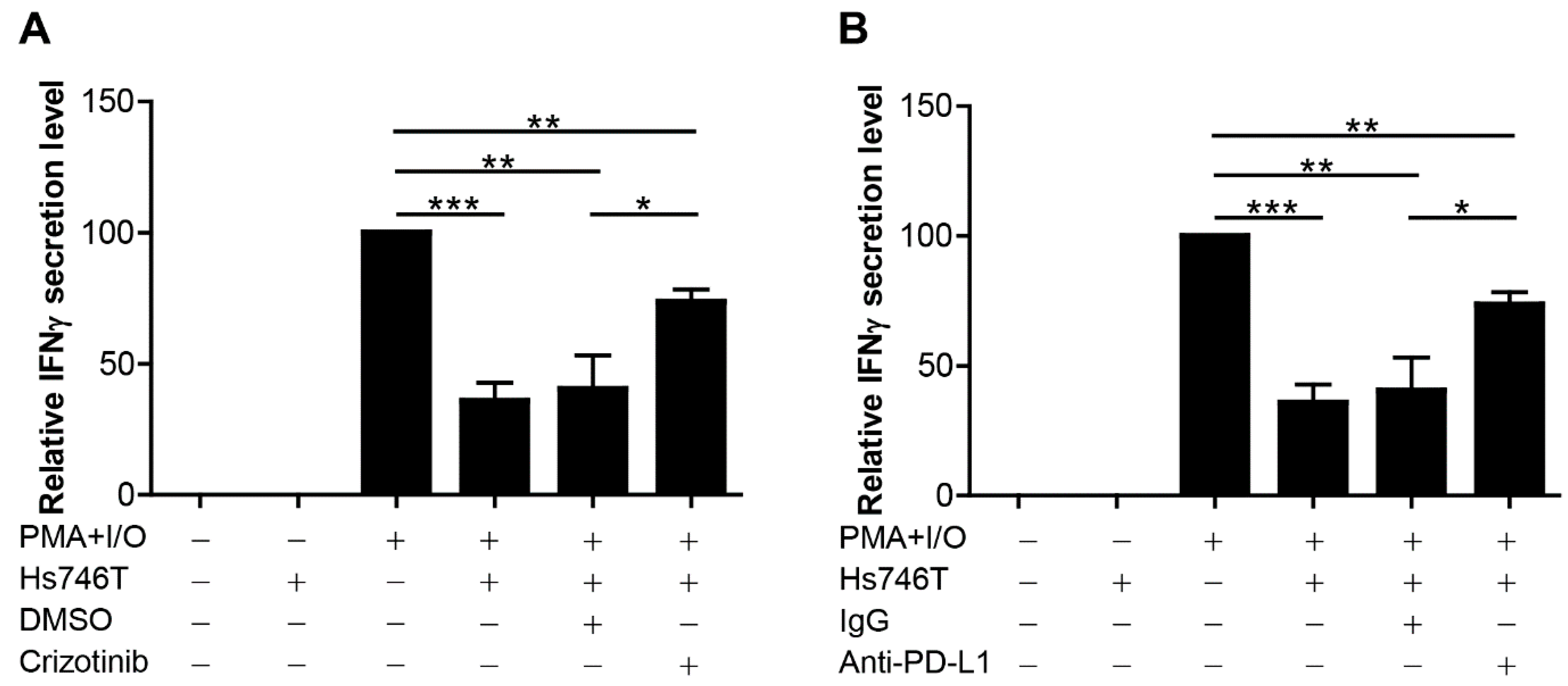
2.3. MET-Overexpressing Cancer Cells Suppress the Function of Immune Cells
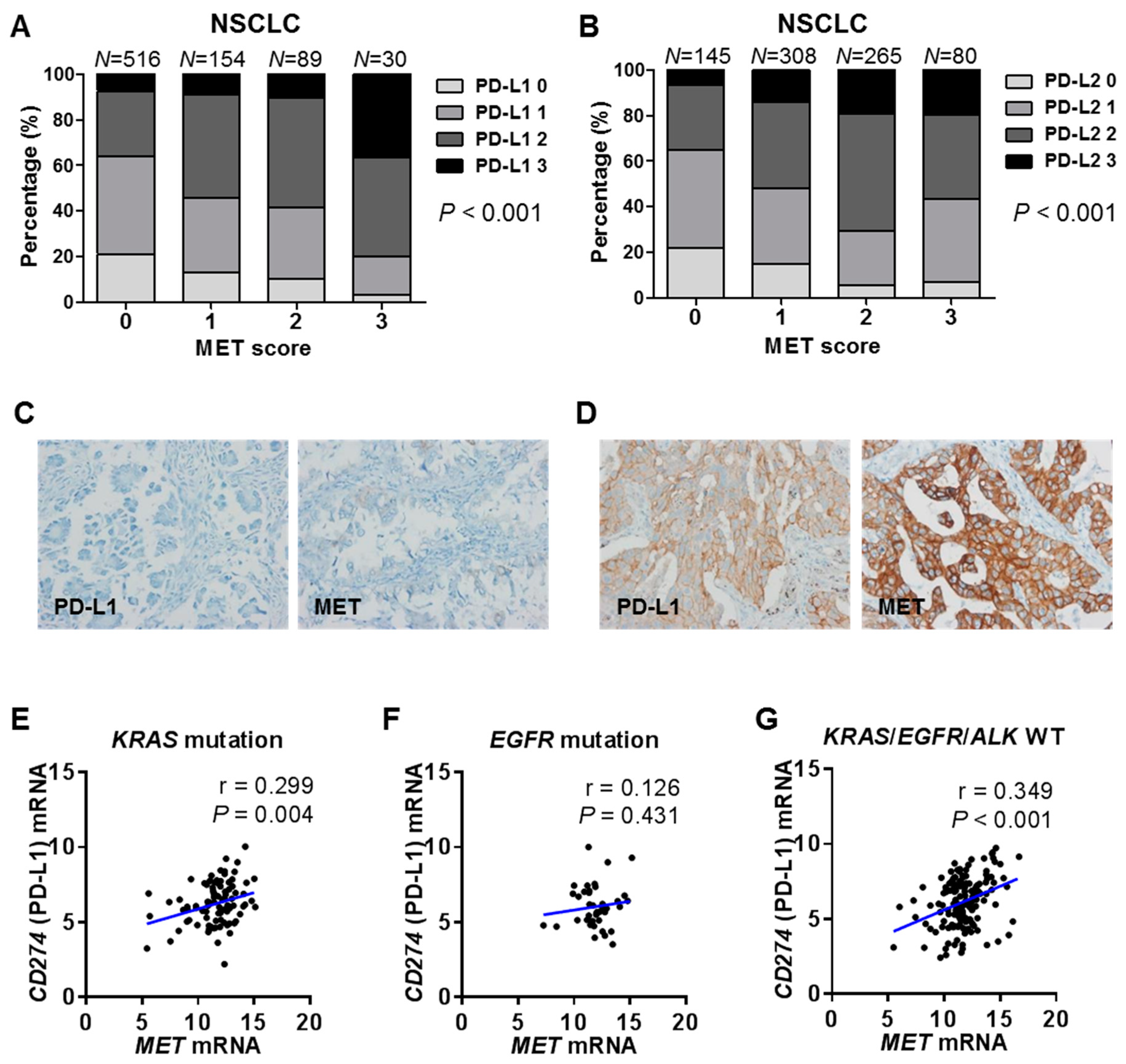
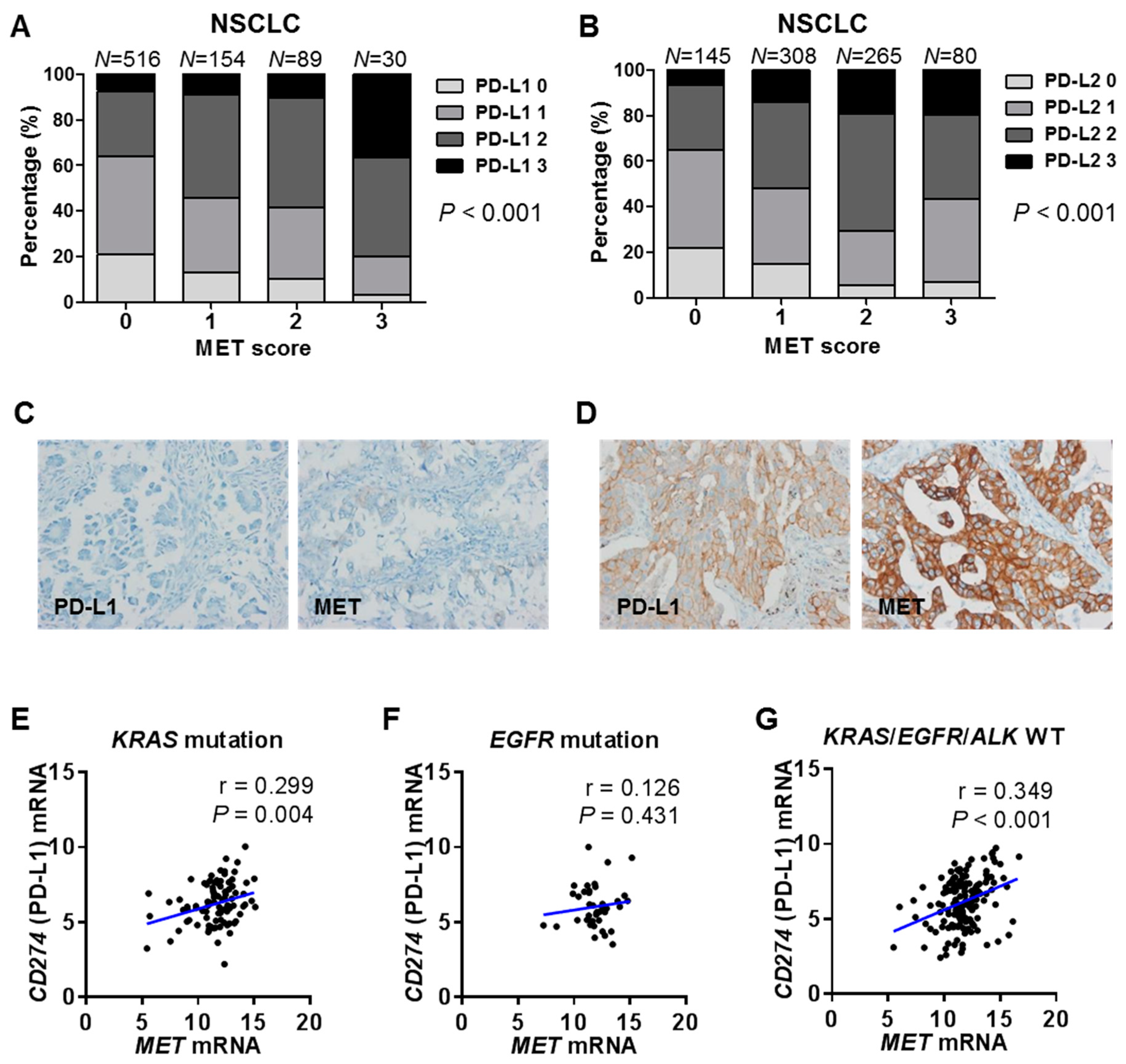
2.4. Correlations between MET and PD-1 Ligand Expression in Human Cancer Tissues and Cell Lines
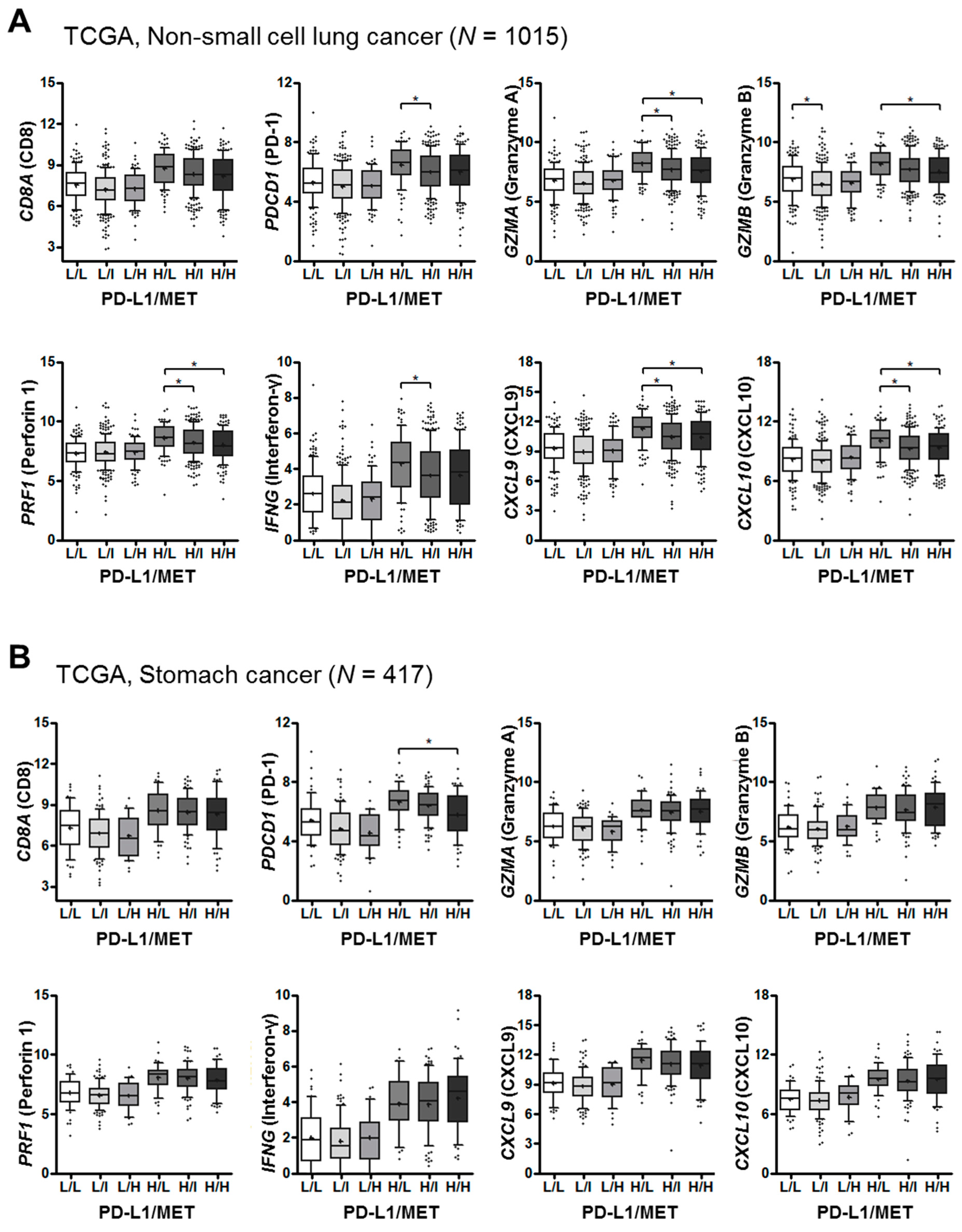
2.5. Landscape of MET, PD-L1 and T-cell Effector Molecule Expression in Human Cancers
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Small Interfering RNA (siRNA) Transfections
4.3. Oligonucleotide Microarray Analysis
4.4. Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
4.5. Flow Cytometry
4.6. Western Blot
4.7. Co-Culture of Human Peripheral Blood Mononuclear Cells (Pbmcs) and Tumor Cells
4.8. Enzyme-Linked Immunosorbent Assay (ELISA) for IFNγ
4.9. Immunohistochemistry (IHC)
4.10. Cancer Cell Line Encyclopedia (CCLE) and the Cancer Genome Atlas (TCGA) Data Analyses
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CCLE | Cancer Cell Line Encyclopedia |
| IFNγ | interferon-γ |
| IHC | immunohistochemistry |
| NSCLC | non-small cell lung cancer |
| PBMC | peripheral blood mononuclear cells |
| PD-1 | programmed cell death-1 |
| PD-L1 | programmed cell death-1-ligand 1 |
| PD-L2 | programmed cell death-1-ligand 2 |
| PMA | phorbol 12-myristate 13-acetate |
| qRT-PCR | quantitative reverse transcriptase–polymerase chain reaction |
| rhHGF | recombinant human hepatocyte growth factor |
| TCGA | The Cancer Genome Atlas |
| TKI | tyrosine kinase inhibitor |
References
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F.; Facchinetti, F.; Haspinger, E.R.; Garassino, M.C.; Trusolino, L.; De Braud, F.; Tiseo, M. Targeting the MET gene for the treatment of non-small-cell lung cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.; Metzenmacher, M.; Goetz, M.; Savvidou, N.; Darwiche, K.; Aigner, C.; Herold, T.; Eberhardt, W.E.; Skiba, C.; Hense, J.; et al. MET Expression in Advanced Non-Small-Cell Lung Cancer: Effect on Clinical Outcomes of Chemotherapy, Targeted Therapy, and Immunotherapy. Clin. Lung Cancer 2018, 19, e441–e463. [Google Scholar] [CrossRef] [PubMed]
- Schildhaus, H.U.; Schultheis, A.M.; Ruschoff, J.; Binot, E.; Merkelbach-Bruse, S.; Fassunke, J.; Schulte, W.; Ko, Y.D.; Schlesinger, A.; Bos, M.; et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res. 2015, 21, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2009, 4, 5–11. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Janne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef]
- Liu, X.; Jia, Y.; Stoopler, M.B.; Shen, Y.; Cheng, H.; Chen, J.; Mansukhani, M.; Koul, S.; Halmos, B.; Borczuk, A.C. Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J. Clin. Oncol. 2016, 34, 794–802. [Google Scholar] [CrossRef]
- Kwon, D.; Koh, J.; Kim, S.; Go, H.; Kim, Y.A.; Keam, B.; Kim, T.M.; Kim, D.W.; Jeon, Y.K.; Chung, D.H. MET exon 14 skipping mutation in triple-negative pulmonary adenocarcinomas and pleomorphic carcinomas: An analysis of intratumoral MET status heterogeneity and clinicopathological characteristics. Lung Cancer 2017, 106, 131–137. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of Non-small Cell Lung Carcinoma with poor prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [PubMed]
- Prelaj, A.; Tay, R.; Ferrara, R.; Chaput, N.; Besse, B.; Califano, R. Predictive biomarkers of response for immune checkpoint inhibitors in non-small-cell lung cancer. Eur. J. Cancer 2019, 106, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Richard, C.; Ledys, F.; Klopfenstein, Q.; Joubert, P.; Routy, B.; Truntzer, C.; Gagne, A.; Hamel, M.A.; Guimaraes, C.F.; et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 2018, 119, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Go, H.; Keam, B.; Kim, M.Y.; Nam, S.J.; Kim, T.M.; Lee, S.H.; Min, H.S.; Kim, Y.T.; Kim, D.W.; et al. Clinicopathologic analysis of programmed cell death-1 and programmed cell death-ligand 1 and 2 expressions in pulmonary adenocarcinoma: Comparison with histology and driver oncogenic alteration status. Mod. Pathol. 2015, 28, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Koh, J.; Kim, S.; Go, H.; Jeon, Y.K.; Chung, D.H. Clinicopathological analysis of PD-L1 and PD-L2 expression in pulmonary squamous cell carcinoma: Comparison with tumor-infiltrating T cells and the status of oncogenic drivers. Lung Cancer 2015, 88, 24–33. [Google Scholar] [CrossRef]
- Maroun, C.R.; Rowlands, T. The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014, 142, 316–338. [Google Scholar] [CrossRef]
- Saigi, M.; Alburquerque-Bejar, J.J.; Mc, L.-F.A.; Pereira, C.; Pros, E.; Romero, O.A.; Baixeras, N.; Esteve-Codina, A.; Nadal, E.; Brambilla, E.; et al. MET-oncogenic and JAK2-inactivating alterations are independent factors that affect regulation of PD-L1 expression in lung cancer. Clin. Cancer Res. 2018, 24, 4579–4587. [Google Scholar] [CrossRef]
- Shin, S.J.; Jeon, Y.K.; Kim, P.J.; Cho, Y.M.; Koh, J.; Chung, D.H.; Go, H. Clinicopathologic Analysis of PD-L1 and PD-L2 Expression in Renal Cell Carcinoma: Association with Oncogenic Proteins Status. Ann. Surg. Oncol. 2016, 23, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Guo, J.; Wen, X.; Ding, G.; Li, B.; Dong, B.; Feng, Q.; Li, S.; Zhang, J.; Cheng, X.; et al. Analysis of PD1, PDL1, PDL2 expression and T cells infiltration in 1014 gastric cancer patients. Oncoimmunology 2018, 7, e1356144. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; Kim, D.W.; Koh, J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Change in PD-L1 Expression After Acquiring Resistance to Gefitinib in EGFR-Mutant Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2016, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Andersen, M.N.; Jakobsen, K.R.; Madsen, A.T.; Sorensen, B.S. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget 2017, 8, 68221–68229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, V.; Chiriaco, C.; Modica, C.; Acquadro, A.; Cortese, M.; Galimi, F.; Perera, T.; Gammaitoni, L.; Aglietta, M.; Comoglio, P.M.; et al. Met inhibition revokes IFNgamma-induction of PD-1 ligands in MET-amplified tumours. Br. J. Cancer 2019, 120, 527–536. [Google Scholar] [CrossRef]
- Li, H.; Li, C.W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.C.; Hsu, J.M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Koh, J.; Jang, J.Y.; Keam, B.; Kim, S.; Kim, M.Y.; Go, H.; Kim, T.M.; Kim, D.W.; Kim, C.W.; Jeon, Y.K.; et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1alpha and STAT3. Oncoimmunology 2016, 5, e1108514. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type (n) | MET | PD-L1 | r *,1 | p1 | PD-L2 | r *,2 | p2 |
|---|---|---|---|---|---|---|---|
| Mean (Median) | Mean (Median) | Mean (Median) | |||||
| HNSCC (n = 519) | 11.05 (11.12) | 6.36 (6.35) | 0.11 | 0.012 | 6.65 (6.63) | 0.174 | 0.000 |
| NSCLC (n = 1015) | 11.40 (11.45) | 6.24 (6.07) | 0.219 † | 0.000 | 6.50 (6.54) | 0.181 | 0.000 |
| Lung ADC (n = 513) | 11.66 (11.84) | 6.05 (5.98) | 0.401 | 0.000 | 6.46 (6.50) | 0.266 | 0.000 |
| Lung SqCC (n = 502) | 11.13 (11.24) | 6.43 (6.22) | 0.144 | 0.001 | 6.53 (6.57) | 0.085 | 0.058 |
| Eso cancer (n = 184) | 11.53 (11.47) | 5.43 (5.35) | 0.02 | 0.792 | 5.46 (5.41) | −0.046 | 0.535 |
| Stomach cancer (n = 415) | 11.31 (11.25) | 5.35 (5.30) | 0.137 | 0.005 | 5.84 (6.00) | 0.015 | 0.754 |
| Colon ADC (n = 380) | 11.77 (11.74) | 4.28 (4.14) | −0.098 | 0.056 | 4.44 (4.56) | −0.169 | 0.001 |
| HCC (n = 371) | 11.57 (11.62) | 3.22 (3.20) | −0.085 | 0.102 | 4.37 (4.28) | −0.107 | 0.039 |
| Breast cancer (n = 1095) | 8.31 (8.43) | 4.43 (4.38) | 0.238 | 0.000 | 5.96 (6.07) | 0.383 | 0.000 |
| Kidney RCC (n = 534) | 12.60 (12.59) | 5.35 (5.38) | 0.19 | 0.000 | 6.40 (6.45) | 0.108 | 0.013 |
| Bladder cancer (n = 407) | 10.56 (10.59) | 4.69 (4.32) | 0.385 | 0.000 | 4.96 (4.99) | 0.355 | 0.000 |
| GBM (n = 157) | 7.43 (7.40) | 4.88 (4.88) | 0.226 | 0.004 | 6.63 (6.81) | 0.237 | 0.003 |
| DLBCL (n = 48) | 7.03 (6.83) | 6.30 (6.05) | 0.12 | 0.415 | 8.25 (8.19) | −0.045 | 0.762 |
| Mesothelioma (n = 87) | 11.70 (12.13) | 4.76 (4.39) | 0.022 | 0.842 | 6.70 (6.55) | −0.079 | 0.468 |
| Sarcoma (n = 258) | 8.47 (8.23) | 4.11 (4.06) | 0.124 | 0.047 | 7.06 (7.29) | 0.122 | 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, H.K.; Kim, S.; Kwon, D.; Koh, J.; Kim, Y.A.; Kim, K.; Chung, D.H.; Jeon, Y.K. MET Receptor Tyrosine Kinase Regulates the Expression of Co-Stimulatory and Co-Inhibitory Molecules in Tumor Cells and Contributes to PD-L1-Mediated Suppression of Immune Cell Function. Int. J. Mol. Sci. 2019, 20, 4287. https://doi.org/10.3390/ijms20174287
Ahn HK, Kim S, Kwon D, Koh J, Kim YA, Kim K, Chung DH, Jeon YK. MET Receptor Tyrosine Kinase Regulates the Expression of Co-Stimulatory and Co-Inhibitory Molecules in Tumor Cells and Contributes to PD-L1-Mediated Suppression of Immune Cell Function. International Journal of Molecular Sciences. 2019; 20(17):4287. https://doi.org/10.3390/ijms20174287
Chicago/Turabian StyleAhn, Hyun Kyung, Sehui Kim, Dohee Kwon, Jaemoon Koh, Young A. Kim, Kwangsoo Kim, Doo Hyun Chung, and Yoon Kyung Jeon. 2019. "MET Receptor Tyrosine Kinase Regulates the Expression of Co-Stimulatory and Co-Inhibitory Molecules in Tumor Cells and Contributes to PD-L1-Mediated Suppression of Immune Cell Function" International Journal of Molecular Sciences 20, no. 17: 4287. https://doi.org/10.3390/ijms20174287