Lessons on Differential Neuronal-Death-Vulnerability from Familial Cases of Parkinson’s and Alzheimer’s Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
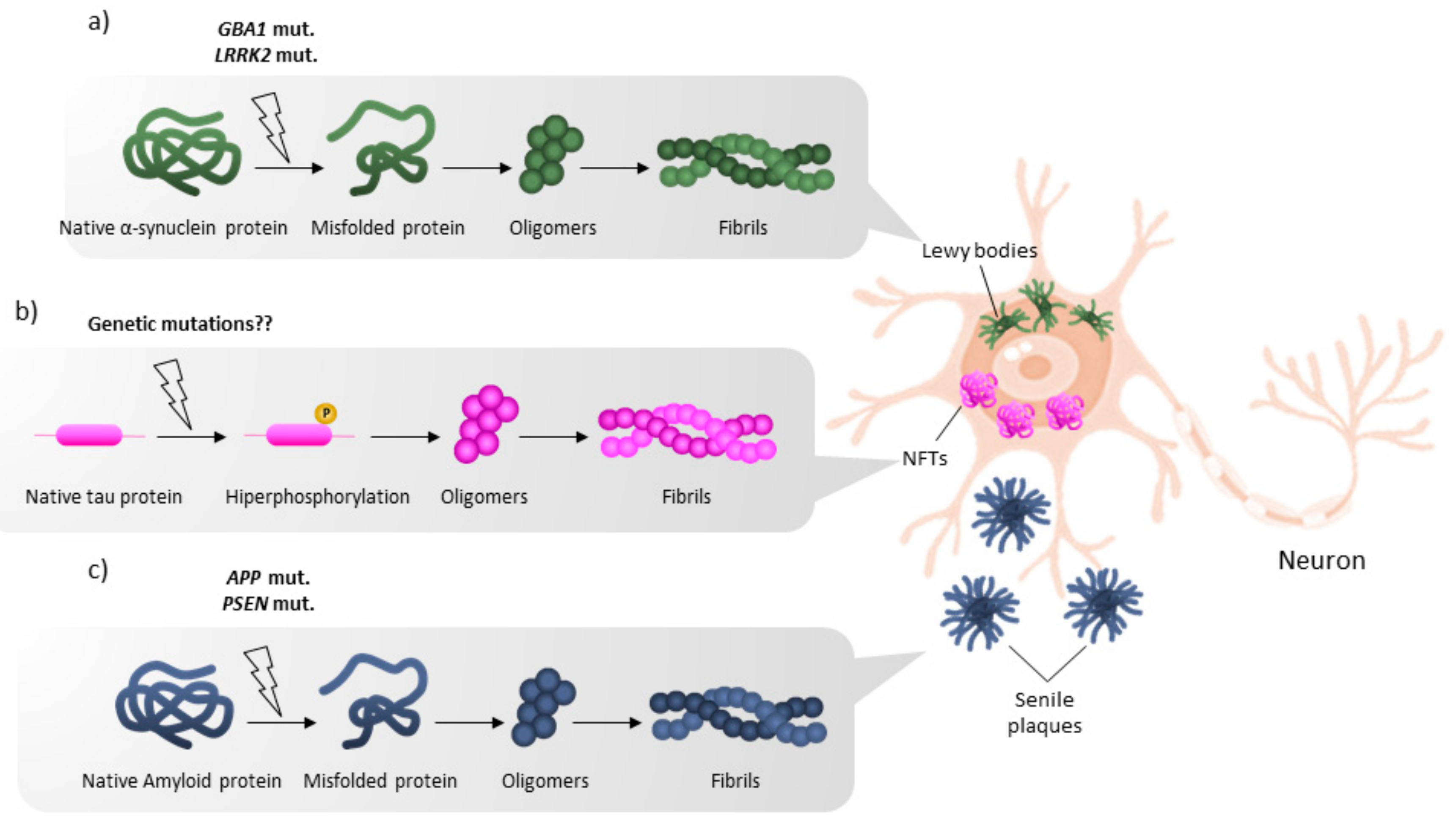
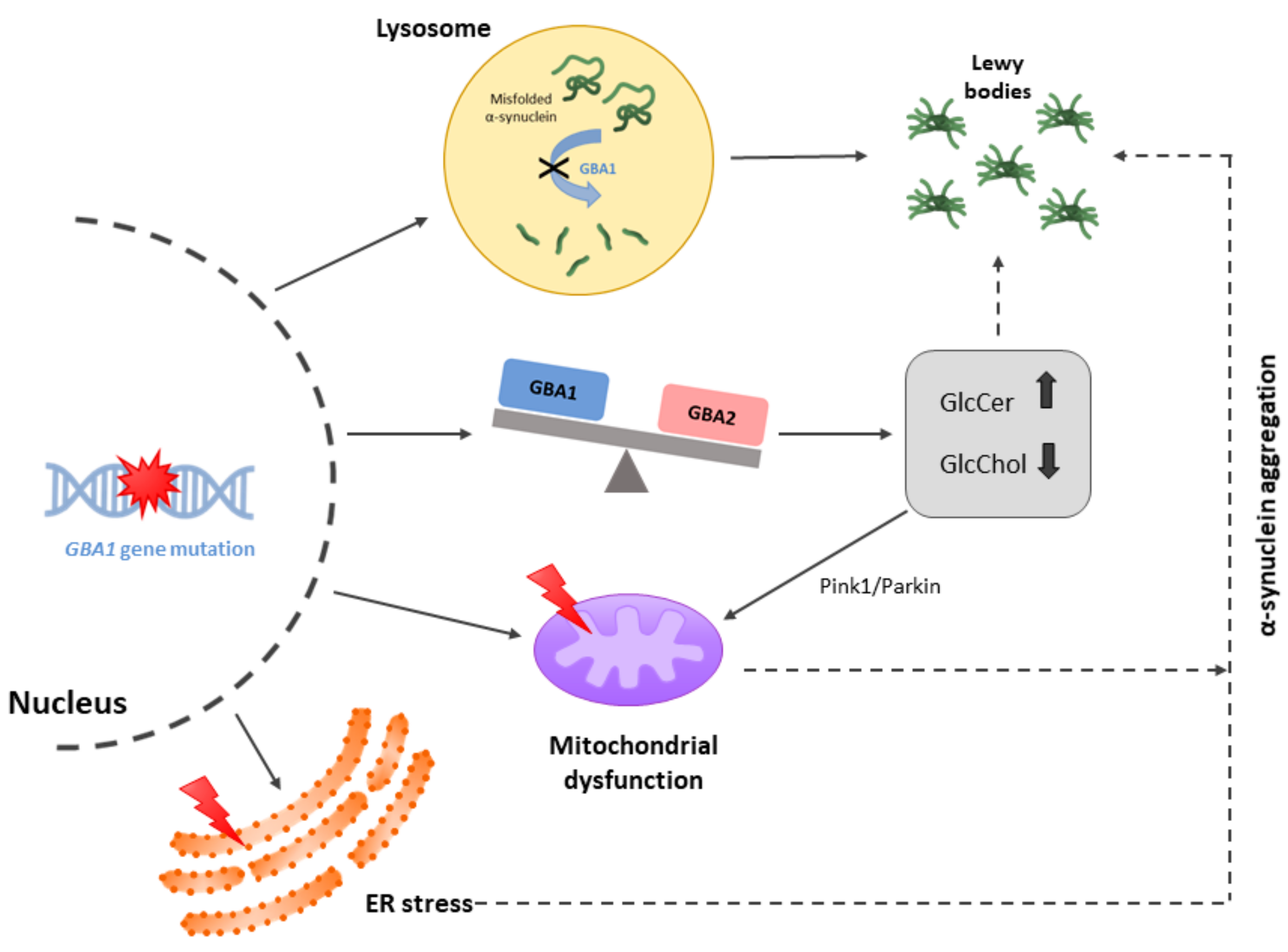
2. Mutations Leading to PD but Not to AD
3. Clues Revealed by Reports on Leucine-Rich Kinase 2 (LRRK2) and Parkin
4. Main Mutations Leading to Inheritable Early-Onset AD but Not to PD
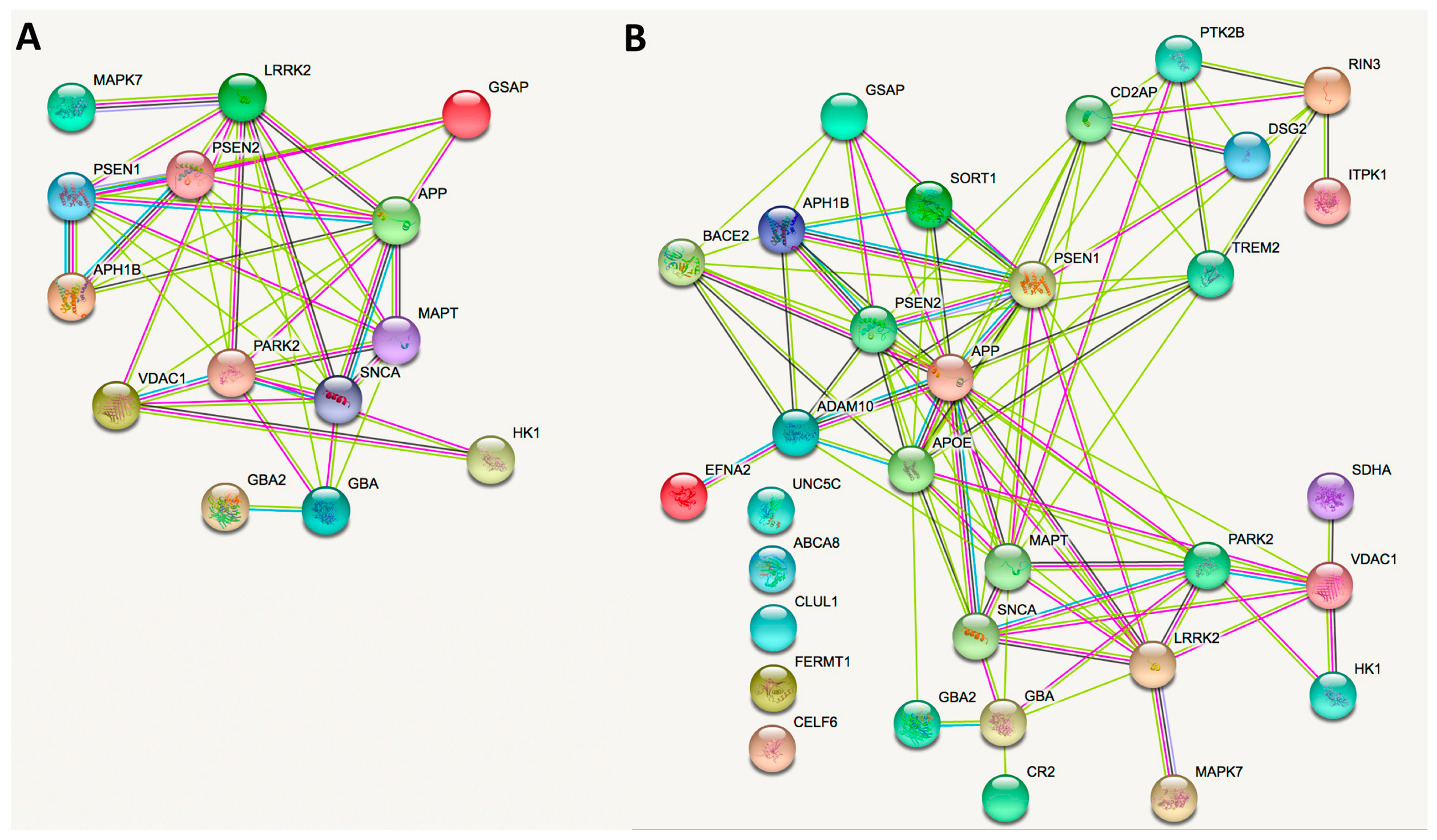
5. Network and Cell Function Analyses
6. Remarks and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AICD | Amyloid intracellular domain |
| APP | Amyloid precursor protein |
| APPSwe | K670M/N671L Swedish mutations |
| CLUL1 | Clusterin |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CR1L | Complement C3b/C4b Receptor 1 like |
| DLB | Dementia with Lewy bodies |
| GBA1 | β-glucocerebrosidase 1 |
| GBA2 | β-glucocerebrosidase 2 |
| GD | Gaucher’s disease |
| GlcChol | β-glucosyl-cholesterol |
| GlcCer | Glucosylceramide |
| LIMP-2 | Lysosomal integral membrane protein-2 |
| LRRK2 | Leucine-rich kinase 2 |
| PD | Parkinson’s disease |
| UNC5C | UNC-5 family of netrin receptors |
| VDAC1 | Voltage-dependent anion-selective channel |
References
- Parkkinen, L.; O’Sullivan, S.S.; Collins, C.; Petrie, A.; Holton, J.L.; Revesz, T.; Lees, A.J. Disentangling the relationship between lewy bodies and nigral neuronal loss in Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 277–286. [Google Scholar] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cookson, M.R. Proteomics; applications in familial Parkinson’s disease. J. Neurochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Mallett, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; O’Reilly, É.J.; Hughes, K.C.; Gao, X.; Schwarzschild, M.A.; Hannan, M.T.; Betensky, R.A.; Ascherio, A. Integration of risk factors for Parkinson disease in 2 large longitudinal cohorts. Neurology 2018, 90, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Boscher, E.; Husson, T.; Quenez, O.; Laquerrière, A.; Marguet, F.; Cassinari, K.; Wallon, D.; Martinaud, O.; Charbonnier, C.; Nicolas, G.; et al. Copy Number Variants in miR-138 as a Potential Risk Factor for Early-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Ramirez Aguilar, L.; Acosta-Uribe, J.; Giraldo, M.M.; Moreno, S.; Baena, A.; Alzate, D.; Cuastumal, R.; Aguillón, D.; Madrigal, L.; Saldarriaga, A.; et al. Genetic origin of a large family with a novel PSEN1 mutation (Ile416Thr). Alzheimers Dement. 2019, 15, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, L. Un cas de maladie de Gaucher de la dulte avec syndrome de Raynaud, pigmentation, et rigidite du type extrapyr-ajidal aux membres inferieurs. Ann. Med. 1939, 45, 57–70. [Google Scholar]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef]
- Bae, E.-J.; Yang, N.Y.; Lee, C.; Lee, H.-J.; Kim, S.; Sardi, S.P.; Lee, S.-J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef]
- Vanderjagt, D.J.; Fry, D.E.; Glew, R.H. Human glucocerebrosidase catalyses transglucosylation between glucocerebroside and retinol. Biochem. J. 1994, 300, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Sánchez-Arias, J.A.; Navarro, G.; Lanciego, J.L. Glucocerebrosidase Mutations and Synucleinopathies. Potential Role of Sterylglucosides and Relevance of Studying Both GBA1 and GBA2 Genes. Front. Neuroanatomy 2018, 12. [Google Scholar] [CrossRef]
- Dopeso-Reyes, I.G.; Sucunza, D.; Rico, A.J.; Pignataro, D.; Marín-Ramos, D.; Roda, E.; Rodríguez-Pérez, A.I.; Labandeira-García, J.L.; Lanciego, J.L. Glucocerebrosidase expression patterns in the non-human primate brain. Brain Struct. Funct. 2018, 223, 343–355. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of -Glucocerebrosidase Reduces Pathological -Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Schapira, A.H.V.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 2016, 524, 1236–1258. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Paisan Ruiz, C.; Crawley, A.; Malkani, R.; Werner, J.; Gwinn-Hardy, K.; Dickson, D.; Wavrant DeVrieze, F.; Hardy, J.; Singleton, A. The dardarin G2019S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci. Lett. 2005, 389, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Lang, A.E.; Kawarai, T.; Sato, C.; Salehi-Rad, S.; Fisman, G.K.; Al-Khairallah, T.; St George-Hyslop, P.; Singleton, A.; Rogaeva, E. LRRK2 gene in Parkinson disease: Mutation analysis and case control association study. Neurology 2005, 65, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Roosen, D.A.; Singleton, A.B. Leucine rich repeat kinase knockout (LRRK KO) mouse model: Linking pathological hallmarks of inherited and sporadic Parkinson’s disease. Mov. Disord. 2018, 33, 72. [Google Scholar] [CrossRef] [PubMed]
- Luerman, G.C.; Nguyen, C.; Samaroo, H.; Loos, P.; Xi, H.; Hurtado-Lorenzo, A.; Needle, E.; Stephen Noell, G.; Galatsis, P.; Dunlop, J.; et al. Phosphoproteomic evaluation of pharmacological inhibition of leucine-rich repeat kinase 2 reveals significant off-target effects of LRRK-2-IN-1. J. Neurochem. 2014, 128, 561–576. [Google Scholar] [CrossRef]
- Hindle, S.; Afsari, F.; Stark, M.; Middleton, C.A.; Evans, G.J.O.; Sweeney, S.T.; Elliott, C.J.H. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum. Mol. Genet. 2013, 22, 2129–2140. [Google Scholar] [CrossRef]
- Ng, C.-H.; Mok, S.Z.S.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.-K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.-L. Parkin Protects against LRRK2 G2019S Mutant-Induced Dopaminergic Neurodegeneration in Drosophila. J. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hattori, N.; Mori, H. Genetics of Parkinson’s disease. Biomed. Pharmacother. 1999, 53, 109–116. [Google Scholar] [CrossRef]
- Yi, W.; MacDougall, E.J.; Tang, M.Y.; Krahn, A.I.; Gan-Or, Z.; Trempe, J.-F.; Fon, E.A. The Landscape of Parkin Variants Reveals Pathogenic Mechanisms and Therapeutic Targets in Parkinson’s Disease. Hum. Mol. Genet. 2019. (Epub ahead of print). [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, A.; Garcia-Rozas, P.; Casarejos, M.J.; Pastor, O.; Rodriguez-Navarro, J.A. Lipidomic Alterations in the Mitochondria of Aged Parkin Null Mice Relevant to Autophagy. Front. Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, R.; Ouyang, X.; Ho, M.W.L.; Chia, W.; Yu, F.; Lim, K.-L. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J. Neurosci. 2007, 27, 8563–8570. [Google Scholar] [CrossRef] [PubMed]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-H.; Zheng, H.; Zeng, L.-D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2018, 9, 15132–15143. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lü, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Axelman, K.; Basun, H.; Winblad, B.; Lannfelt, L. A Large Swedish Family with Alzheimer’s Disease with a Codon 670/671 Amyloid Precursor Protein Mutation: A Clinical and Genealogical Investigation. Arch. Neurol. 1994, 51, 1193–1197. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Kamphorst, W.; Heutink, P.; van Swieten, J.C. Tau Pathology in Two Dutch Families with Mutations in the Microtubule-Binding Region of Tau. Am. J. Pathol. 1998, 153, 1359–1363. [Google Scholar] [CrossRef]
- Mullan, M.; Houlden, H.; Windelspecht, M.; Fidani, L.; Lombardi, C.; Diaz, P.; Rossor, M.; Crook, R.; Hardy, J.; Duff, K.; et al. A locus for familial early–onset Alzhelmer’s disease on the long arm of chromosome 14, proximal to the α1–antichymotrypsin gene. Nat. Genet. 1992, 2, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Ancolio, K.; Marambaud, P.; Dauch, P.; Checler, F. Alpha-secretase-derived product of beta-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer’s disease. J. Neurochem. 1997, 69, 2494–2499. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome. Science 1992, 258, 668–671. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.; Haines, J.; Rogaev, E.; Mortilla, M.; Vaula, G.; Pericak-Vance, M.; Foncin, J.-F.; Montesi, M.; Bruni, A.; Sorbi, S.; et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome. Nat. Genet. 1992, 2, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Van Broeckhoven, C.; Backhovens, H.; Cruts, M.; De Winter, G.; Bruyland, M.; Cras, P.; Martin, J.-J. Mapping of a gene predisposing to early–onset Alzheimer’s disease to chromosome 14q24. Nat. Genet. 1992, 2, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Zabetian, C.P.; Lauricella, C.J.; Tsuang, D.W.; Leverenz, J.B.; Schellenberg, G.D.; Payami, H. Analysis of the LRRK2 G2019S mutation in Alzheimer Disease. Arch. Neurol. 2006, 63, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-C.; Zhang, W.; Chua, L.-L.; Chai, C.; Li, R.; Lin, L.; Cao, Z.; Angeles, D.C.; Stanton, L.W.; Peng, J.-H.; et al. Phosphorylation of amyloid precursor protein by mutant LRRK2 promotes AICD activity and neurotoxicity in Parkinson’s disease. Sci. Signal. 2017, 10, eaam6790. [Google Scholar] [CrossRef]
- Same, K.; Ghazi Sherbaf, F.; Aarabi, M.H. New link between Parkinson’s and Alzheimer’s: Research uncovers the role of mutant leucine rich repeat kinase 2 and amyloid precursor protein. Mov. Disord. 2017, 32, 1378–1379. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Cabodevilla, J.F.; Zamarbide, M.; Gómez-Isla, T.; Franco, R.; Pérez-Mediavilla, A. Age-related mitochondrial alterations without neuronal loss in the hippocampus of a transgenic model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-beta. J. Alzheimers Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, I.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Relevance for Alzheimer’s disease and levo-dopa-induced dyskinesia. Brain. Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernández-Suárez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Zamarbide, M.; Gil-Bea, F.J.; Bannenberg, P.; Martínez-Pinilla, E.; Sandoval, J.; Franco, R.; Pérez-Mediavilla, A. Maternal imprinting on cognition markers of wild type and transgenic Alzheimer’s disease model mice. Sci. Rep. 2018, 8, 6434. [Google Scholar] [CrossRef]
- Breitner, J.C.S.; Welsh, K.A. Genes and Recent Developments in the Epidemiology of Alzheimer’s Disease and Related Dementia. Epidemiol. Rev. 1995, 17, 39–47. [Google Scholar] [CrossRef]
- Cruts, M.; Hendriks, L.; Van Broeckhoven, C. The presenilin genes: A new gene family involved in Alzheimer disease pathology. Hum. Mol. Genet. 1996, 5, 1449–1455. [Google Scholar] [CrossRef]
- Duggan, S.P.; McCarthy, J. V Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell. Signal. 2016, 28, 1–11. [Google Scholar] [CrossRef]
- Lee, S.H.; Lutz, D.; Mossalam, M.; Bolshakov, V.Y.; Frotscher, M.; Shen, J. Presenilins regulate synaptic plasticity and mitochondrial calcium homeostasis in the hippocampal mossy fiber pathway. Mol. Neurodegener. 2017, 12, 48. [Google Scholar] [CrossRef]
- Tacik, P.; Sanchez-Contreras, M.; Rademakers, R.; Dickson, D.W.; Wszolek, Z.K. Genetic Disorders with Tau Pathology: A Review of the Literature and Report of Two Patients with Tauopathy and Positive Family Histories. Neurodegener. Dis. 2016, 16, 12–21. [Google Scholar] [CrossRef]
- Grünblatt, E.; Zander, N.; Bartl, J.; Jie, L.; Monoranu, C.-M.; Arzberger, T.; Ravid, R.; Roggendorf, W.; Gerlach, M.; Riederer, P. Comparison analysis of gene expression patterns between sporadic Alzheimer’s and Parkinson’s disease. J. Alzheimers. Dis. 2007, 12, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Head, D.; Shah, A.R.; Marcus, D.; Mintun, M.; Morris, J.C.; Holtzman, D.M. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann. Neurol. 2009, 65, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical”. Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Wennberg, A.M.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Machulda, M.M.; Petrucelli, L.; Mielke, M.M.; Jack, C.R.; Knopman, D.S.; et al. The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol. Aging 2019, 77, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Y. Mutations in the ATP13A2 Gene and Parkinsonism: A Preliminary Review. Biomed Res. Int. 2014, 2014, 1–9. [Google Scholar]
- Van Kampen, J.M.; Baranowski, D.C.; Robertson, H.A.; Shaw, C.A.; Kay, D.G. The Progressive BSSG Rat Model of Parkinson’s: Recapitulating Multiple Key Features of the Human Disease. PLoS ONE 2015, 10, e0139694. [Google Scholar] [CrossRef]
- Van Kampen, J.M.; Robertson, H.A. The BSSG rat model of Parkinson’s disease: Progressing towards a valid, predictive model of disease. EPMA J. 2017, 8, 261–271. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, P.; Orgaz, L.; Fuentes, J.M.; Vicario, C.; Moratalla, R. Cholesterol and multilamellar bodies: Lyosomal dysfunction in GBA -Parkinson disease. Autophagy 2018, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Hirabayashi, Y. A novel function for glucocerebrosidase as a regulator of sterylglucoside metabolism. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.; Minnich, A.; Davignon, J. Apolipoprotein E, synaptic plasticity and Alzheimer’s disease. Ann. Med. 1995, 27, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Xue-Shan, Z.; Juan, P.; Qi, W.; Zhong, R.; Li-hong, P.; Zhi-han, T.; Zhi-Sheng, J.; Gui-xue, W.; Lu-Shan, L. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin. Chim. Acta 2016, 456, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Rebeck, G. The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk. Int. J. Mol. Sci. 2018, 20, 63. [Google Scholar] [CrossRef]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Navarro, G.; Martínez-Pinilla, E. Lessons on Differential Neuronal-Death-Vulnerability from Familial Cases of Parkinson’s and Alzheimer’s Diseases. Int. J. Mol. Sci. 2019, 20, 3297. https://doi.org/10.3390/ijms20133297
Franco R, Navarro G, Martínez-Pinilla E. Lessons on Differential Neuronal-Death-Vulnerability from Familial Cases of Parkinson’s and Alzheimer’s Diseases. International Journal of Molecular Sciences. 2019; 20(13):3297. https://doi.org/10.3390/ijms20133297
Chicago/Turabian StyleFranco, Rafael, Gemma Navarro, and Eva Martínez-Pinilla. 2019. "Lessons on Differential Neuronal-Death-Vulnerability from Familial Cases of Parkinson’s and Alzheimer’s Diseases" International Journal of Molecular Sciences 20, no. 13: 3297. https://doi.org/10.3390/ijms20133297
APA StyleFranco, R., Navarro, G., & Martínez-Pinilla, E. (2019). Lessons on Differential Neuronal-Death-Vulnerability from Familial Cases of Parkinson’s and Alzheimer’s Diseases. International Journal of Molecular Sciences, 20(13), 3297. https://doi.org/10.3390/ijms20133297