Abstract
Transcripts of many enzymes involved in base excision repair (BER) undergo extensive alternative splicing, but functions of the corresponding alternative splice variants remain largely unexplored. In this review, we cover the studies describing the common alternatively spliced isoforms and disease-associated variants of DNA glycosylases, AP-endonuclease 1, and DNA polymerase beta. We also discuss the roles of alternative splicing in the regulation of their expression, catalytic activities, and intracellular transport.
1. Introduction
Base excision repair (BER) is the predominant and conserved pathway that corrects small DNA lesions derived from oxidation, deamination, and alkylation (reviewed in [1,2,3,4,5]). BER is initiated by a DNA glycosylase that removes damaged or mismatched nucleobase, leaving an apurinic/apyrimidinic site (AP site). At least 11 human DNA glycosylases are known, each recognizing one or a few related lesions, but also demonstrating overlapping specificities. AP sites are further cleaved by an AP endonuclease (APEX1 in humans) yielding a 3′ hydroxyl and a 5′ deoxyribose phosphate moiety (dRP). Alternatively, bifunctional DNA glycosylases not only excise damaged bases but follow with damaged-strand nicking by β-elimination. Regardless of the nick origin, it can then be processed by either short-patch (where a single nucleotide is replaced) or long-patch (where 2 to 13 nucleotides are replaced) BER. In the former case, after the AP site cleavage, DNA repair polymerase (DNA polymerase beta, or Pol β, in human cells) removes the dRP group, through its dRP lyase activity, and fills the gap. In the latter sub-pathway, Pol β and possibly other DNA polymerases displace the nicked strand in the 5′→3′ direction, and the resulting flap is then excised by FEN1 endonuclease. BER plays a crucial role in maintaining genomic stability (recently reviewed in [6]). A number of mutations and single-nucleotide polymorphisms were shown to be associated with an increased risk of human diseases including cancer (reviewed in [7,8,9]).
Alternative splicing of pre-mRNA has been shown to affect about 95% of human genes [10,11,12], and is involved in the regulation of normal physiological functions as well as in pathologic processes [13,14]. Alternative splicing increases functional diversity and provides additional regulatory opportunities. In particular, protein isoforms generated by alternative splicing could be different in their catalytic capacity, subcellular localization, or protein–protein interactions. In addition, alternative splicing could negatively regulate gene expression (e.g., regulated unproductive splicing and translation realized through the nonsense-mediated decay of alternatively spliced mRNA isoforms harboring a premature termination codon) [15].
Many BER enzymes are associated with several mRNA and protein variants from a single gene. The transcript diversity of BER enzymes resulting from alternative splicing has yet to be explored. Little is known about the effect of common alternative splicing events on the BER enzymes activities and functions. In this review, we address alternative splicing of key BER enzymes: DNA glycosylases, APEX1, and Pol β. We summarize the studies describing common alternatively spliced isoforms of BER enzymes and their known disease-associated variants. We also discuss the possible roles of alternative splicing in the regulation of BER enzyme expression and activities.
2. Isoforms of DNA Glycosylases and Apurinic/Apyrimidinic Endonuclease
2.1. Uracil–DNA Glycosylase (UNG)
Uracil–DNA glycosylase was the first discovered human DNA glycosylase and the first one for which the existence of different mRNA and protein isoforms was confirmed [16,17,18,19,20,21,22,23]. The UNG gene contains seven exons and two alternative transcription initiation sites that generate two mRNA and two protein isoforms (often called UNG1 and UNG2). Exon 1 is only included in the UNG2 mRNA isoform, while exon 2 is the first exon of UNG1 and incorporates part of intron 1 where the UNG1 transcription start is located [19,20,23,24,25] (Figure 1). Direct promoter activity mapping suggests that there might be yet another untranslated exon upstream of exon 1, important for cell cycle-dependent regulation involving the E2F family transcription factors [26,27].
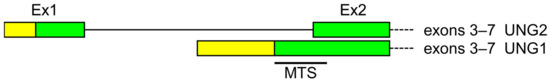
Figure 1.
Organization of the alternative transcription initiation in the human UNG gene. Untranslated regions are yellow, coding sequences are green. MTS—mitochondrial targeting sequence.
Both UNG isoforms share the 269 amino acids (aa) long catalytic domain and possess nearly identical enzymatic activity, removing uracil from any context in DNA [28,29,30]. The isoform-specific functions entailed by the unique N-termini (35 aa in UNG1, 44 aa in UNG2) are related primarily to intracellular trafficking, protein–protein interactions, and regulatory post-translational modification.
The UNG1 protein isoform carries a classical N-terminal mitochondrial targeting sequence (MTS) and is imported into mitochondria, whereas the UNG2 isoform is localized to the nucleus due to a complex nuclear localization signal (NLS) split between its unique N-terminus and the catalytic domain common with UNG1 [19,23,31]. Interestingly, while the mouse Ung gene has a similar structure and also produces two protein isoforms, mouse UNG1 protein has an N-terminus quite different from that of human UNG1 and is sorted both to mitochondria and to nuclei [32,33].
Under oxidative stress, UNG1 forms a disulfide bond through its Cys5 with a major mitochondrial antioxidant protein peroxiredoxin 3, which protects UNG1 from damage and reduces mitochondrial DNA oxidation [34]. The nuclear isoform, UNG2, is cell-cycle regulated, targeted for degradation in the late S phase through ubiquitylation dependent on phosphorylation within the isoform-specific N-terminal region [35,36,37]. On the other hand, UNG2 is phosphorylated at Thr6, and its dephosphorylation by protein phosphatase 1D partially suppresses the enzyme activity [38]. In addition, the N-terminal tail of UNG2 interacts with proliferating cell nuclear antigen (PCNA) clamp and replication protein A (RPA) [39], raising the possibility of replication-coupled uracil repair, which makes sense considering massive amounts of uracil misincorporated into DNA during the replication [40].
2.2. Thymine–DNA Glycosylase (TDG)
TDG, which removes an array of pyrimidine oxidation/deamination products, is now considered to be a major component of the epigenetic active cytosine demethylation system rather than a bona fide DNA repair enzyme [41,42]. Human TDG gene contains ten exons and generates two mRNA isoforms, one of which produces a full-length, extensively characterized protein, whereas the second encodes a truncated protein lacking part of the active site and is likely non-functional [24,43,44,45,46]. The mouse Tdg gene expresses two mRNAs with different incorporation of exon 1 parts that have different translation initiation sites [47,48,49,50,51]. The polypeptides, TDGa and TDGb, appear to behave identically with respect to their ability to bind SUMO-1 but were not compared otherwise [51].
2.3. Single-Strand-Selective Monofunctional Uracil-DNA Glycosylase 1 (SMUG1)
SMUG1 has enzymatic activity and substrate specificity similar to UNG and is regarded as a backup for UNG [30,52]. Its gene contains six exons, with multiple transcription start sites, cryptic splicing sites within exons 1 and 6, and skipping of exons 2–5 producing 32 known mRNA isoforms, the largest number among all DNA glycosylases [24,44,53,54,55,56] (Table 1). However, they encode only five different protein isoforms, of which only one has been biochemically characterized [52,54], whereas all the rest are predicted to lack important parts of the catalytic domain.

Table 1.
Experimentally confirmed and predicted mRNA and protein isoforms of major BER genes.
2.4. Methyl-Binding Domain-Containing Protein 4 (MBD4)
MBD4 is an enzyme that removes cytosine and 5-methylcytosine deamination products from CpG dinucleotides and has a methyl-binding domain directing it to CpG-islands; it may have both DNA repair and epigenetic demethylation functions [41,58,59]. The human MBD4 gene possesses eight exons; skipping of exon 3 and alternative splicing donor sites in exon 7 produce five mRNA and five protein isoforms [44,58,59,60,61,62,63]. Two protein isoforms (3 and 4) terminate prematurely and lack portions of the catalytic domain. In addition to the major full-length protein isoform 1, a splice variant (isoform 5) skipping exons that code for the methyl-binding domain but possessing an intact glycosylase domain have been characterized [63]. The protein displayed robust uracil-excising activity, coincident with the truncated MBD4 used in many biochemical studies [64,65,66]. Two probable pathogenic splice variant mutations of MBD4 associated with glioblastoma and uveal melanoma [67] were reported in the Human Gene Mutation Database (HGMD) [68] (Table 2).
Table 2.
Pathogenic splice variants of MBD4, NTHL1, OGG1, NEIL1, and NEIL2 genes.
2.5. Endonuclease III-Like Protein (NTHL1)
NTHL1 is the main human glycosylase for the repair of oxidized pyrimidines [74,75]. The contains six exons, with alternative transcription start sites in exons 1 and 2, and exon 3 skipped in one of three known mRNA variants [25,44,74,75,76,77,78,79]. Only one of three resulting protein isoforms has intact catalytic domain and can be considered functional. The translation initiation site of the NTHL1 open reading frame is ambiguous, since the first 16 sequence positions contain three methionines and four predicted mRNA capping sites. However, the enhanced green fluorescent protein (EGFP)-tagged NTHL1 starting at any of these three Met positions are localized identically, both to nucleus and to mitochondria [79,80,81]. Remarkably, an abnormal splicing-causing mutation in intron 4 of the NTHL1 gene associated with several tumor phenotypes (colonic adenocarcinoma, bladder carcinoma, intradermal nevi, meningioma, multiple seborrheic keratoses, basal cell carcinoma, multiple colorectal adenomas, squamous cell carcinoma, and invasive ductal carcinoma) was reported (Table 2).
2.6. 8-Oxoguanine–DNA Glycosylase (OGG1)
OGG1 is responsible for removal of an abundant pre-mutagenic oxidative lesion, 8-oxoguanine (oxoG). OGG1 together with another DNA glycosylase, MUTYH, and 8-oxodGTPase NUDT1 (MTH1) constitute a so-called GO system that controls oxoG at pre- and post-replicative levels [82,83]. The human OGG1 gene contains eight exons plus one small, irregularly-used exon 7.5 [84,85,86]. The intron between exons 7 and 8 contains nearly a half of calmodulin kinase 1 (CAMK1) gene transcribed in the opposite direction. Alternative splicing generates two groups of OGG1 mRNA isoforms. OGG1 group 1 isoforms (1a–1e) include exons 1 to 7, whereas exon 8 substitutes for exon 7 in group 2 mRNAs (2a–2h); within those groups, isoforms differ from each other by the use of some internal exons and alternative splice sites [44,84,85,87,88,89,90,91,92,93,94,95,96,97] (Figure 2A). Interestingly, group 2 isoforms have been reported in primates only, and even extensively-annotated transcriptomes of other animals, such as mice, chicken, or zebrafish, show no signs of them.
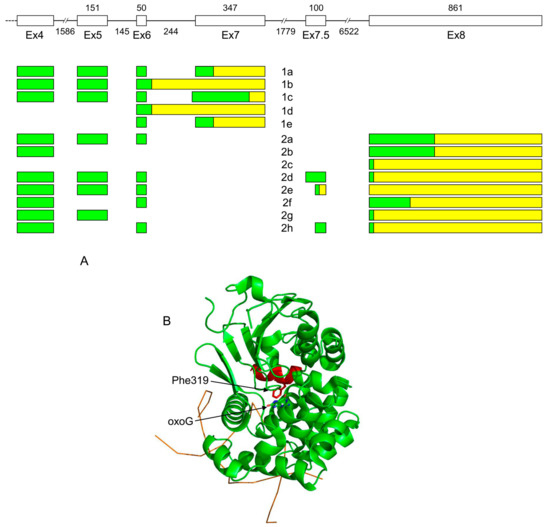
Figure 2.
(A) Organization of exons 4–8 and alternative splicing products of the human OGG1 gene. (B) Structure of OGG1-1a protein (PDB ID 1EBM, [98]). DNA backbone is shown schematically as an orange line. The protein part encoded by exon 7 and thus missing in OGG1-1b and all group 2 isoforms are colored red. Phe319 forms a wall of the enzyme active site and stacks against the oxoG base; these interactions have to be remodeled in OGG1 group 2. Untranslated regions are yellow, coding sequences are green.
Following the mRNA differences, the protein isoforms of OGG1 fall in two groups with different C-termini. Since group 2 protein isoforms contains only the N-terminal MTS, while group 1 isoforms also contains the NLS in its C-terminal part, it was initially believed that these groups comprise mitochondrial and nuclear proteins, respectively [95], and groups 1 and 2 are still often referred to as nuclear and mitochondrial OGG1 isoforms, respectively. However, immunocytochemical localization of OGG1-1a detected it both in the nucleus and in the cytoplasm in a speckled pattern characteristic of mitochondrial proteins, whereas OGG1-1b, -1c, and -2a were excluded from the nucleus and found only in mitochondria [80,99]. Moreover, high-resolution microscopy indicates that OGG1-1a resides in the mitochondrial matrix, associated with mtDNA in the nucleoid [99]. The N-terminal target peptide is required for mitochondrial localization of OGG1 regardless of the presence of the NLS, and is necessary to complement the mitochondrial function under the oxidative stress conditions [80,99]. The G12E somatic mutation found in renal clear cell carcinomas abolishes mitochondrial localization of OGG1-2a without affecting the activity of OGG1-1a [100].
OGG1-1a protein has been extensively biochemically characterized. After the structure of OGG1-1a was determined [98], it became evident that its active site is partly built from the polypeptide region unique for this isoform, including Phe319 that stacks against oxoG in the recognition pocket (Figure 2B). Therefore, other isoforms, if active, have to use some other mode of oxoG recognition. In line with these structural considerations, the recombinant OGG1-2a protein, which deviates from OGG1-1a starting at position 317, was reported to lack enzymatic activity [101]. Paradoxically, OGG1-1b, which also lacks Phe319, was reported to possess oxoG-excising activity at the level comparable with OGG1-1a [102]. Overall, the question of enzymatic activity of various OGG1 protein isoforms remains unsolved.
Despite the lack of activity of OGG1-2a isoform, the oxoG excision activity encoded by OGG1 is found in mitochondria [103,104]. In principle, participation of OGG1-2a still cannot be excluded, since the protein expressed intracellularly in Escherichia coli might not fully reflect the properties of the protein after its unfolding and refolding by the mitochondrial import system [105,106]. However, given that group 1 isoforms are also detected in mitochondria by Western blotting and immunocytochemistry [80,99,101], it is very likely that OGG1-1a is responsible for the oxoG removal activity there, and group 2 isoforms may play some other role. Intriguingly, OGG1-1a targeted to mitochondria by additional SOD2 signal peptide makes cells more resistant to oxidative stress even if OGG1 carries inactivating mutations in the active site; this ability apparently depends on blocking oxidant-induced decreases in mitochondrial aconitase activity [107].
In the yeast two-hybrid system, OGG1-2a interacts with NADH:ubiquinone oxidoreductase subunit B10, an integral part of the NADH dehydrogenase (ubiquinone) complex residing in the mitochondrial inner membrane [108]. Selective knockdown of both group 1 and group 2 by targeting exons 7 and 8, respectively, sensitizes cells to oxidative stress, impairs the respiration, and increases the oxoG content in their mitochondrial DNA, while overexpression of OGG1-2a but not of OGG1-1a increases oxoG removal activity in mitochondria and protects cells from oxidative damage [108,109].
It is not clear yet whether the minor mRNA isoforms (1b–1e, 2b–2h) are translated. Western blotting with monoclonal antibodies directed against the putative NLS reveals several immunoreactive OGG1 protein bands [96,110], suggesting that some nuclear isoforms do appear in human cells. Cells under oxidative stress have been reported to accumulate shorter OGG1 polypeptides that are either minor isoforms or produced by apoptosis-related proteolysis [111,112].
Some pathologic processes can change the balance of OGG1 isoforms, although no cause–effect relations have been established. Group 2 isoforms are increased in the substantia nigra in Parkinson’s disease patients [113]. OGG1-1a and -1c transcripts are significantly upregulated, whereas OGG1-1b is downregulated in Alzheimer’s disease patients in comparison with age-matched controls [114].
The HGMD contains three splice-site altered OGG1 variants. One of them is reported as a disease-causing variant (colorectal cancer) and two mutations are marked as disease-associated polymorphisms associated with breast cancer (Table 2) [70,71]. A possibly pathogenic mutation OGG1 c.137G>A affecting the last nucleotide in exon 1 was discovered in heterozygote in a colorectal cancer patient who also carried a heterozygous I223V mutation in the coding region of MUTYH (Table 2). The c.137G>A variant led to a complete disappearance of mutant mRNA from the patients’ cells, with no aberrant splicing products present [70,115].
2.7. MutY Homolog (MUTYH)
MUTYH is responsible for removing A mispaired with oxoG, and also can excise several oxidized purine derivatives [116,117]. The MUTYH gene consists of 17 exons [25,44,45,56,118,119,120,121]. Alternative transcription initiation and splicing of the MUTYH pre-mRNA produces at least 13 mRNA isoforms and nine protein isoforms with different 5′-terminal mRNA and N-terminal protein sequences [120,122]. The experimentally identified transcripts form three groups—α, β, and γ—with different transcription initiation sites, and there are multiple instances of exon skipping and alternative splice site use [120]. One source of mRNA variability may be the overlap between exon 1 of MUTYH and exon 1 of TOE1 gene transcribed in the opposite direction under the control of early growth response protein 1 (EGR-1) promoter [55,123]. The major nuclear protein isoform, MUTYHα1, is 546 aa long. The functions of other protein isoforms are unclear. All but one have an intact catalytic domain and are likely to be functional; they may have different catalytic activity and vary in the opposite-base specificity [124], and some of them are probably mitochondrial [80,119,120]. The N-terminal 32-aa peptide also contains a binding site for replication protein A, implying that some isoforms of MUTYH may participate in replication-coupled repair [125]. In mouse cells, three mRNA isoforms are synthesized, two encoding identical polypeptides (MUTYHα) homologous to the major human isoform, while the third uses an alternative translation initiation site and skips one internal exon, resulting in a protein lacking the DNA minor groove-binding motif and likely inactive [122]. In the rat brain, specific mitochondrial isoforms of MUTYH protein have been observed, which are developmentally regulated and induced by respiratory hypoxia in the hippocampus [126,127].
The involvement of MUTYH deficiency in the pathogenesis of colorectal cancer [116,128] spurred interest in possible splice-site mutations in human malignancies. A germline variant, c.892-2A>G, encoding a truncated protein without the NUDIX domain, was discovered in gastric cancer patients; however, its association with cancer risk has not been established due to a small number of cases investigated [129]. Another polymorphism discovered in VMRC-LCD lung cancer cell line, IVS1+5G/C, is located in the first intron and causes incorporation of extra 237 nucleotides in the 5′-UTR of group β isoforms [121]. While it does not change the protein sequence, it appears to reduce the translation efficiency of the carrier mRNAs [121].
The search of MUTYH pathogenic splice variants in the HGMD revealed 19 splice-site mutations reported as a disease-causing mutation, eight splice mutations marked as probable pathological mutation, and one mutation caused reduced translation efficiency of MUTYH transcripts. The majority of MUTYH splice mutations are implicated in MUTYH-associated polyposis and other types of colorectal cancer (Table 3).
Table 3.
Pathogenic splice variants of MUTYH gene.
2.8. Methylpurine–DNA Glycosylase (MPG)
MPG (alias AAG or APNG) excises ring-alkylated purines and several other purine-derived lesions, such as etheno adducts and hypoxanthine [143]. The MPG gene contains six exons that produce three mRNAs and three protein isoforms (MPGa–MPGc) due to alternative transcription initiation and alternative splicing that produces mRNA with one of two alternative first exons [144,145,146]. The protein isoforms are different only in a short N-terminal sequence and are 283–298 aa long [144,145]. All isoforms appear to be ubiquitously expressed, and the respective proteins show identical activity, substrate specificity, and the ability to protect E. coli from alkylation DNA damage when produced ectopically [144,147,148].
2.9. Endonuclease VIII-Like Proteins (NEIL1, NEIL2, and NEIL3)
NEIL1, NEIL2, and NEIL3 are homologs of bacterial endonuclease VIII (Nei); they are involved in the repair of oxidized bases and most likely are needed in special cases, such as repair in non-canonical DNA structures, transcription-coupled repair, or repair in certain cell types or at specific developmental stages [149,150,151]. In humans, NEIL1 gene contains eleven introns, generates four alternatively spliced mRNAs, and encodes four protein isoforms [24,44,55,152,153]. Of these, only one contains no deletions of the essential parts of the protein; the others have not been studied. In mice, in addition to mRNA encoding full-length NEIL1, two splice variants were detected, one containing full intron 4, another including the first 10 nt of intron 1. Both variants also produce truncated proteins, which lack fully or partially the C-terminal DNA-binding domain and possess no catalytic activity [154].
Interestingly, Arabidopsis MMH-1 protein, belonging to a group of plant and fungal H2TH glycosylases that is phylogenetically closest to NEIL1, has multiple isoforms with variable C-termini due to alternative splicing of its pre-mRNA [155,156,157]. Only two variants, MMH-1 and MMH-2, have been characterized biochemically, and only the former had the glycosylase and AP lyase activities, whereas the latter lacks part of the C-terminal domain and is inactive [156].
The NEIL2 gene contains five exons and produces eight mRNA isoforms due to alternative splicing [24,44,53,55,153,158,159]. Three protein isoforms are produced; two of them lack parts of the catalytic domain and are presumably inactive.
The NEIL3 gene contains ten exons. A single mRNA and a single protein isoform are known [44,45,55,62,153,158,160].
One splice variant of NEIL1 reported as disease-associated polymorphism with additional functional evidence was identified but its clinical significance is not obvious [72]. NEIL2 has one splice-site mutation reported as probable pathological splice variant mutation associated with multiple colorectal adenomas [73] (Table 2).
2.10. Apurinic/Apyrimidinic Endonuclease (APEX1)
APEX1 (also known as APE1, HAP1, or Ref-1) catalyzes the next step after DNA glycosylases, hydrolyzing the DNA backbone 5′ to the AP site formed by the DNA glycosylase action [161]. The APEX1 gene contains five exons and produces four mRNA isoforms that differ by using splice donor sites in the untranslated exon 1 and all produce the same polypeptide [44,162,163,164,165,166,167,168,169,170,171,172].
3. Isoforms of DNA Polymerase β
Pol β, belonging to the X family of DNA polymerases, performs most of the gap-filling synthesis in the course of short-patch BER in nuclei [173,174] and in mitochondria [175,176]. In addition to the polymerase activity, Pol β also possesses a 5′-dRP lyase activity [177]. In addition, Pol β is involved in long-patch BER [178,179,180].
Pol β is encoded by a single-copy 34 kb gene located on chromosome 8p11 [181]. The POLB gene consists of 14 exons ranging in size from 50 to 233 bp and 13 introns (Figure 3) [173,181,182,183]. The major Pol β isoform is a 39 kDa protein consisting of 335 amino acid residues. Pol β is folded into two distinct domains, each associated with a specific functional activity: the N-terminal 8 kDa domain (encoded by exons 1–4) shows the dRP lyase activity and the C-terminal 31 kDa domain (encoded by exons 5–14) possesses the DNA-polymerase activity [184,185].
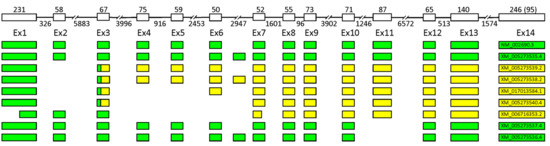
Figure 3.
The schematic intron–exon organization of the human POLB gene and its common alternative splicing products. Untranslated regions are yellow, coding sequences are green.
POLB is a housekeeping gene expressed at low levels throughout the cell cycle [186]. Remarkably, the POLB gene is highly conserved in mammals (e.g., 99% among primates and 87% between rat and human), but the frequency of POLB splice variants in human cells is extremely high, reaching half total POLB transcripts [181,187,188,189,190,191,192,193,194]. About 60 splice variants of POLB were detected by sequencing of mRNAs and RT-PCR in different types of human normal tissues and cancer cells (Supplementary Materials). Much lower POLB splice variants levels were reported in other mammalian and non-mammalian species [188,191,194], suggesting that the POLB splice variants pattern observed in humans is not evolutionarily preserved and might play some adaptive functions. The majority of the POLB splice variants contain a premature terminating codon and are not able to produce a functional protein (Supplementary Materials).
Common POLB splice variants include deletions of exons 2, 4, 5, 6, 11, and 13 (∆), separately and in various combinations (Figure 3). Deletion of exon 2 (∆2) is the most frequent alternative splice event found almost in all cell types and tissues studied and conserved among primates [188,192]. Deletion of exon 2 leads to a frameshift and premature transcription termination in exon 3, resulting in production of truncated protein containing the first 26 amino acid residues [187,190]. The translated protein of 29 kDa should possess single-strand DNA binding and dRP-lyase activities. However, overexpression of this splice isoform does not rescue hypersensitivity to the cytotoxic effect of methyl methanesulfonate (MMS) in Pol β null cells and does not affect alkylating agent sensitivity and BER capability of Pol β proficient cells [192]. It was shown that the Ex2Δ mRNA is not translated in vitro [191] and its translation product is non-detectable in cell extracts (even in the presence of proteasome inhibitors), suggesting that deletion of exon 2 gives rise to a non-coding RNA transcript that could either represent unproductive and nonfunctional splice variant of POLB or modulate target mRNAs [192]. The possible role of Ex2Δ mRNA as a post-transcriptional regulator is supported by its localization at the sites of the active translation on polyribosomes, polyadenylation, and longer half-life [192]. A two-fold increase in the Ex2Δ POLB expression level does not induce any change in the levels of POLB mRNA but other genes could possibly be regulated.
The only confirmed protein isoform of Pol β detected in cells by Western-blot, along with the wild type Pol β, is a 36 kDa protein derived from translation of Ex11Δ mRNA [195,196,197]. Exon 11 encodes amino acid residues 208–236 and its deletion leads to the loss of 29 amino acids from the palm domain of Pol β. In many studies, the Ex11Δ variant was observed in tumors but not in the corresponding normal tissues [188,195,196,197,198,199,200,201], while no correlation with a gastric cancer phenotype was observed [192]. Cells expressing the Ex11Δ variant demonstrate decreased survival following exposure to alkylating agents [201,202,203]. The Ex11Δ protein suppresses BER reactions in transfected cells and cell extracts suggesting that this isoform acts as a dominant negative regulator of Pol β-dependent BER [201,202,204]. Expression of the Ex11Δ variant in mammary glands promotes carcinogenesis in transgenic mice after N-methyl-N-nitrosourea (MNU) treatment [203]. The Ex11Δ variant is capable of DNA gap-filling synthesis almost as effective as wild-type Pol β, and it binds a number of BER proteins: XRCC1, PARP1, APEX1, and TAF1D [204]. It is remarkable that binary complex of the Ex11Δ isoform and XRCC1 cannot perform the gap-filling reaction and has enhanced affinity to gapped DNA, suggesting that the Ex11Δ-Pol β-XRCC1 inhibits Pol β by substrate competition [204].
Deletion of several exons starting from exon 11 (∆11–13) are also common in tumors and cancer cell lines [188,190,191,198,200,205,206] and is possibly caused by mutations in the exon–intron junction [205]. HeLa cells transfected with the Ex11-13∆ variant express protein of 26.5 kDa lacking amino acids 208–304 of the catalytic core of Pol β, and display high sensitivity to alkylating agents and moderate sensitivity to UV and H2O2 [206]. This isoform is likely deficient in the DNA polymerase activity, [207] but retains the dRP lyase and DNA binding activities [208], and could act as a dominant negative mutant of Pol β. POLB splice variant Ex4–6,11–13∆ is associated with ovarian cancer [205].
Other common splice variants are retention of introns 6, 9, and 11 (Σ) [188,189,190,191,192,193,196,199,207,209]. Introns 6 and 9 were named exons α and β, respectively [187,191]. Retention of intron 6 does not affect the POLB reading frame and could possibly lead to production of a 42 kDa protein containing an additional 35 residues in the α-spiral region of the fingers domain [187]. The Σ exon α isoform produced in E. coli retains the catalytic activity but demonstrates reduced solubility [187]. There is no evidence of the existence of this isoform at the protein level in human cells yet.
Interestingly, it was observed that suppression of the nonsense-mediated decay pathway in human fibroblasts increases the splice variant frequency and amount of POLB isoforms containing a premature stop codon [190]. Therefore, part of the unproductive POLB splice variants could play a role in the nonsense-mediated mRNA decay mechanism.
4. Conclusions
Transcripts encoding BER enzymes undergo extensive alternative splicing that serves diverse purposes. It leads to production of proteins with grossly different catalytic capacity compared with their major annotated isoform (as OGG1-1 and -2), regulates their intracellular transport (as in UNG1 and UNG2), or provides a post-transcriptional regulation mechanism. Such regulation can include production of truncated proteins that act as dominant negative factors and inhibit the activity of the major isoform by substrate competition (e.g., Pol β Ex11Δ isoform), production of non-coding RNAs modulating target mRNAs (e.g., Pol β Ex2Δ isoform), or production of non-functional RNA isoforms degraded by nonsense-mediated decay. At the same time, the functions of many alternative splice variants of BER proteins remain largely unexplored.
In some cases, splicing-affecting mutations in BER genes have been directly shown to be pathogenic. This is best illustrated by the example of MUTYH, where a number of splicing-affecting mutations, both germline and somatic, have been discovered in human tumors. Although their frequency is apparently lower compared to the well-characterized missense and truncating MUTYH mutations causative of colorectal polyposis and cancer, there is also a growing number of observations that aberrantly spliced transcripts can be produced even in the absence of mutations through splicing dysregulation in cancer cells [210,211]. Presently, the research field of normal and pathogenic splicing in base excision repair is ripe for discovery.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/20/13/3279/s1.
Author Contributions
Writing—original draft preparation, E.O.B., R.F.K., A.V.M. and D.O.Z.; writing—review and editing, A.V.M. and D.O.Z.; visualization, E.O.B. and D.O.Z.; funding acquisition, A.V.M. and D.O.Z.
Funding
This research was funded by the Russian Foundation for Basic Research grant 17-00-00264 (to A.V.M.), the Russian Science Foundation for Basic Research grant 17-00-00263 (to R.F.K), and the Russian Science Foundation 17-14-01190 (to D.O.Z., DNA glycosylases and AP endonucleases). Partial salary support from the Russian Ministry of Science and Education is recognized (6.5773.2017/6.7 and AAAA-A17-117020210023-1 to D. O. Z.). Publication fees were paid by an intramural program of SB RAS Institute for Chemical Biology and Fundamental Medicine.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| BER | base excision repair |
| AP | an abasic site |
| APEX1 | AP endonuclease 1 |
| dRP | 5′ deoxyribose phosphate moiety |
| Pol β | DNA polymerase beta |
| UNG | uracil–DNA glycosylase |
| TDG | thymine–DNA glycosylase |
| SMUG1 | single-strand-selective monofunctional uracil-DNA glycosylase 1 |
| MBD4 | methyl-binding domain-containing protein 4 |
| NTHL1 | endonuclease III-like protein |
| OGG1 | 8-oxoguanine–DNA glycosylase |
| MUTYH | MutY homolog |
| MPG | methylpurine–DNA glycosylase |
| NEIL1, NEIL2, NEIL3 | endonuclease VIII-like proteins |
| PCNA | proliferating cell nuclear antigen |
| RPA | replication protein A |
| MTS | mitochondrial targeting sequence |
| NLS | nuclear localization signal |
| MMS | methyl methanesulfonate |
References
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, A.V.; Bondarenko, K.A.; Makarova, A.V. Non-bulky lesions in human DNA: The ways of formation, repair, and replication. Acta Naturae 2017, 9, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.V.; Makarova, A.V.; Miftakhova, R.R.; Zharkov, D.O. Base excision DNA repair deficient cells: From disease models to genotoxicity sensors. Curr. Pharm. Des. 2019, 25, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, D.; Dalal, S.; Sweasy, J.B. Is there a link between DNA polymerase β and cancer? Cell Cycle 2004, 3, 998–1001. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Marsden, C.G.; Dragon, J.A.; Wallace, S.S.; Sweasy, J.B. Base excision repair variants in cancer. Methods Enzymol. 2017, 591, 119–157. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The size of the human proteome: The width and depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Brooks, A.N.; Soergel, D.A.; Meng, Q.; Brenner, S.E. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv. Exp. Med. Biol. 2007, 623, 190–211. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.; Wittwer, C.U. Uracil DNA-glycosylase from HeLa cells: General properties, substrate specificity and effect of uracil analogs. Nucleic Acids Res. 1981, 9, 2599–2613. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.C.; Aasland, R.; Wittwer, C.U.; Krokan, H.E.; Helland, D.E. Molecular cloning of human uracil-DNA glycosylase, a highly conserved DNA repair enzyme. EMBO J. 1989, 8, 3121–3125. [Google Scholar] [CrossRef]
- Wittwer, C.U.; Bauw, G.; Krokan, H.E. Purification and determination of the NH2-terminal amino acid sequence of uracil-DNA glycosylase from human placenta. Biochemistry 1989, 28, 780–784. [Google Scholar] [CrossRef]
- Slupphaug, G.; Markussen, F.-H.; Olsen, L.C.; Aasland, R.; Aarsæther, N.; Bakke, O.; Krokan, H.E.; Helland, D.E. Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 1993, 21, 2579–2584. [Google Scholar] [CrossRef]
- Haug, T.; Skorpen, F.; Lund, H.; Krokan, H.E. Structure of the gene for human uracil-DNA glycosylase and analysis of the promoter function. FEBS Lett. 1994, 353, 180–184. [Google Scholar] [CrossRef]
- Caradonna, S.; Ladner, R.; Hansbury, M.; Kosciuk, M.; Lynch, F.; Muller, S. Affinity purification and comparative analysis of two distinct human uracil-DNA glycosylases. Exp. Cell Res. 1996, 222, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Haug, T.; Skorpen, F.; Kvaløy, K.; Eftedal, I.; Lund, H.; Krokan, H.E. Human uracil-DNA glycosylase gene: Sequence organization, methylation pattern, and mapping to chromosome 12q23–q24.1. Genomics 1996, 36, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.-I.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shen, P.; Thiyagarajan, S.; Lin, S.; Palm, C.; Horvath, R.; Klopstock, T.; Cutler, D.; Pique, L.; Schrijver, I.; et al. Identification of rare DNA variants in mitochondrial disorders with improved array-based sequencing. Nucleic Acids Res. 2011, 39, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Haug, T.; Skorpen, F.; Aas, P.A.; Malm, V.; Skjelbred, C.; Krokan, H.E. Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res. 1998, 26, 1449–1457. [Google Scholar] [CrossRef]
- Muller-Weeks, S.; Balzer, R.J.; Anderson, R.; Caradonna, S. Proliferation-dependent expression of nuclear uracil-DNA glycosylase is mediated in part by E2F-4. DNA Repair 2005, 4, 183–190. [Google Scholar] [CrossRef]
- Bharati, S.; Krokan, H.E.; Kristiansen, L.; Otterlei, M.; Slupphaug, G. Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res. 1998, 26, 4953–4959. [Google Scholar] [CrossRef]
- Parikh, S.S.; Mol, C.D.; Slupphaug, G.; Bharati, S.; Krokan, H.E.; Tainer, J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef]
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef]
- Otterlei, M.; Haug, T.; Nagelhus, T.A.; Slupphaug, G.; Lindmo, T.; Krokan, H.E. Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 1998, 26, 4611–4617. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, P.C.; Yee, H.A.; Winkfein, R.J.; van de Sande, J.H. The mouse uracil-DNA glycosylase gene: Isolation of cDNA and genomic clones and mapping ung to mouse chromosome 5. Gene 1997, 189, 175–181. [Google Scholar] [CrossRef]
- Nilsen, H.; Rosewell, I.; Robins, P.; Skjelbred, C.F.; Andersen, S.; Slupphaug, G.; Daly, G.; Krokan, H.E.; Lindahl, T.; Barnes, D.E. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell 2000, 5, 1059–1065. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, Y.; Gong, Y.; Zhang, W.; Liu, C.; Wang, Q.; Deng, H. Hydrogen peroxide mediated mitochondrial UNG1-PRDX3 interaction and UNG1 degradation. Free Radic. Biol. Med. 2016, 99, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Muller-Weeks, S.; Mastran, B.; Caradonna, S. The nuclear isoform of the highly conserved human uracil-DNA glycosylase is an Mr 36,000 phosphoprotein. J. Biol. Chem. 1998, 273, 21909–21917. [Google Scholar] [CrossRef]
- Fischer, J.A.; Muller-Weeks, S.; Caradonna, S. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair 2004, 3, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.A.; Muller-Weeks, S.; Caradonna, S.J. Fluorodeoxyuridine modulates cellular expression of the DNA base excision repair enzyme uracil-DNA glycosylase. Cancer Res. 2006, 66, 8829–8837. [Google Scholar] [CrossRef]
- Lu, X.; Bocangel, D.; Nannenga, B.; Yamaguchi, H.; Appella, E.; Donehower, L.A. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol. Cell 2004, 15, 621–634. [Google Scholar] [CrossRef]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J. 1999, 18, 3834–3844. [Google Scholar] [CrossRef]
- Andersen, S.; Heine, T.; Sneve, R.; König, I.; Krokan, H.E.; Epe, B.; Nilsen, H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis 2005, 26, 547–555. [Google Scholar] [CrossRef]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Schomacher, L.; Niehrs, C. DNA repair and erasure of 5-methylcytosine in vertebrates. BioEssays 2017, 39, 1600218. [Google Scholar] [CrossRef] [PubMed]
- Sard, L.; Tornielli, S.; Gallinari, P.; Minoletti, F.; Jiricny, J.; Lettieri, T.; Pierotti, M.A.; Sozzi, G.; Radice, P. Chromosomal localizations and molecular analysis of TDG gene-related sequences. Genomics 1997, 44, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Mammalian Gene Collection (MGC) Program Team. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [CrossRef] [PubMed]
- Brandenberger, R.; Wei, H.; Zhang, S.; Lei, S.; Murage, J.; Fisk, G.J.; Li, Y.; Xu, C.; Fang, R.; Guegler, K.; et al. Transcriptome characterization elucidates signaling networks that control human ES cell growth and differentiation. Nat. Biotechnol. 2004, 22, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Fortna, A.; Kim, Y.; MacLaren, E.; Marshall, K.; Hahn, G.; Meltesen, L.; Brenton, M.; Hink, R.; Burgers, S.; Hernandez-Boussard, T.; et al. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004, 2, e207. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, L.; Gallinari, P.; Gariboldi, M.; Manenti, G.; Pierotti, M.A.; Jiricny, J.; Dragani, T.A. Genetic mapping of thymine DNA glycosylase (Tdg) gene and of one pseudogene in the mouse. Mamm. Genome 1996, 7, 909–910. [Google Scholar] [CrossRef] [PubMed]
- Neddermann, P.; Gallinari, P.; Lettieri, T.; Schmid, D.; Truong, O.; Hsuan, J.J.; Wiebauer, K.; Jiricny, J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1996, 271, 12767–12774. [Google Scholar] [CrossRef] [PubMed]
- Um, S.; Harbers, M.; Benecke, A.; Pierrat, B.; Losson, R.; Chambon, P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1998, 273, 20728–20736. [Google Scholar] [CrossRef]
- Hardeland, U.; Steinacher, R.; Jiricny, J.; Schär, P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002, 21, 1456–1464. [Google Scholar] [CrossRef]
- Takahashi, H.; Hatakeyama, S.; Saitoh, H.; Nakayama, K.I. Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J. Biol. Chem. 2005, 280, 5611–5621. [Google Scholar] [CrossRef] [PubMed]
- Visnes, T.; Doseth, B.; Pettersen, H.S.; Hagen, L.; Sousa, M.M.L.; Akbari, M.; Otterlei, M.; Kavli, B.; Slupphaug, G.; Krokan, H.E. Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Yoshitomo-Nakagawa, K.; Maruyama, K.; Suyama, A.; Sugano, S. Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene 1997, 200, 149–156. [Google Scholar] [CrossRef]
- Haushalter, K.A.; Stukenberg, P.T.; Kirschner, M.W.; Verdine, G.L. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 1999, 9, 174–185. [Google Scholar] [CrossRef]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Coulombe-Huntington, J.; Kang, S.; Sheynkman, G.M.; Hao, T.; Richardson, A.; Sun, S.; Yang, F.; Shen, Y.A.; Murray, R.R.; et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 2016, 164, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Thibaud-Nissen, F.; Souvorov, A.; Murphy, T.; DiCuccio, M.; Kitts, P. Eukaryotic genome annotation pipeline. In The NCBI Handbook, 2nd ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2018; pp. 133–156. [Google Scholar]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef]
- Bellacosa, A.; Cicchillitti, L.; Schepis, F.; Riccio, A.; Yeung, A.T.; Matsumoto, Y.; Golemis, E.A.; Genuardi, M.; Neri, G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl. Acad. Sci. USA 1999, 96, 3969–3974. [Google Scholar] [CrossRef]
- Hendrich, B.; Abbott, C.; McQueen, H.; Chambers, D.; Cross, S.; Bird, A. Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm. Genome 1999, 10, 906–912. [Google Scholar] [CrossRef]
- Tomarev, S.I.; Wistow, G.; Raymond, V.; Dubois, S.; Malyukova, I. Gene expression profile of the human trabecular meshwork: NEIBank sequence tag analysis. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2588–2596. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yamashita, R.; Shirota, M.; Sakakibara, Y.; Chiba, J.; Mizushima-Sugano, J.; Nakai, K.; Sugano, S. Sequence comparison of human and mouse genes reveals a homologous block structure in the promoter regions. Genome Res. 2004, 14, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.M.; Baker, R.D.; Bader, S.; Dunlop, M.G.; Nicholl, I.D. The identification of a novel alternatively spliced form of the MBD4 DNA glycosylase. Oncol. Rep. 2007, 17, 111–116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, P.; Qiu, C.; Sohail, A.; Zhang, X.; Bhagwat, A.S.; Cheng, X. Mismatch repair in methylated DNA: Structure and activity of the mismatch-specific thymine glycosylase domain of methyl-CpG-binding protein MBD4. J. Biol. Chem. 2003, 278, 5285–5291. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Zhang, X.; Cheng, X. Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: Structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 8276–8284. [Google Scholar] [CrossRef] [PubMed]
- Moréra, S.; Grin, I.; Vigouroux, A.; Couvé, S.; Henriot, V.; Saparbaev, M.; Ishchenko, A.A. Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res. 2012, 40, 9917–9926. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Fiévet, A.; Gardrat, S.; Barnhill, R.L.; Popova, T.; Servois, V.; Rampanou, A.; Mouton, A.; et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 2018, 9, 1866. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.; Castellsagué, E.; Bah, I.; van Kempen, L.C.; Foulkes, W.D. Biallelic NTHL1 mutations in a woman with multiple primary tumors. N. Engl. J. Med. 2015, 373, 1985–1986. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Massdorf, T.; Sykora, H.; Kerscher, M.; Holinski-Feder, E. First evidence for digenic inheritance in hereditary colorectal cancer by mutations in the base excision repair genes. Eur. J. Cancer 2011, 47, 1046–1055. [Google Scholar] [CrossRef]
- Ali, K.; Mahjabeen, I.; Sabir, M.; Mehmood, H.; Kayani, M.A. OGG1 Mutations and risk of female breast cancer: Meta-analysis and experimental data. Dis. Markers 2015, 2015. [Google Scholar] [CrossRef]
- Dallosso, A.R.; Dolwani, S.; Jones, N.; Jones, S.; Colley, J.; Maynard, J.; Idziaszczyk, S.; Humphreys, V.; Arnold, J.; Donaldson, A.; et al. Inherited predisposition to colorectal adenomas caused by multiple rare alleles of MUTYH but not OGG1, NUDT1, NTH1 or NEIL 1, 2 or 3. Gut 2008, 57, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Colley, J.; Jones, S.; Dallosso, A.R.; Maynard, J.H.; Humphreys, V.; Dolwani, S.; Sampson, J.R.; Cheadle, J.P. Rapid recognition of aberrant dHPLC elution profiles using the Transgenomic Navigator™ software. Hum. Mutat. 2005, 26, 165. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, T.P.; Chaung, W.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J. Biol. Chem. 1997, 272, 6733–6740. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Biswas, T.; Roy, R.; Izumi, T.; Boldogh, I.; Kurosky, A.; Sarker, A.H.; Seki, S.; Mitra, S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III: Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem. 1998, 273, 21585–21593. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, T.P.; Boorstein, R.J.; Kung, H.C.; Bolton, P.H.; Xing, D.; Cunningham, R.P.; Teebor, G.W. Purification of a mammalian homologue of Escherichia coli endonuclease III: Identification of a bovine pyrimidine hydrate-thymine glycol DNA-glycosylase/AP lyase by irreversible cross linking to a thymine glycol-containing oligodeoxynucleotide. Biochemistry 1996, 35, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Sarker, A.H.; Akiyama, K.; Ikeda, S.; Yao, M.; Tsutsui, K.; Shohmori, T.; Seki, S. Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 1998, 222, 287–295. [Google Scholar] [CrossRef]
- Sarker, A.H.; Ikeda, S.; Nakano, H.; Terato, H.; Ide, H.; Imai, K.; Akiyama, K.; Tsutsui, K.; Bo, Z.; Kubo, K.; et al. Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J. Mol. Biol. 1998, 282, 761–774. [Google Scholar] [CrossRef]
- Luna, L.; Bjørås, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000, 460, 95–104. [Google Scholar] [CrossRef]
- Takao, M.; Aburatani, H.; Kobayashi, K.; Yasui, A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998, 26, 2917–2922. [Google Scholar] [CrossRef]
- Ikeda, S.; Kohmoto, T.; Tabata, R.; Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair 2002, 1, 847–854. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Sakumi, K.; Sakamoto, K.; Tsuchimoto, D.; Tsuzuki, T.; Nakatsu, Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem. 2006, 387, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M.; et al. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar] [PubMed]
- Ishida, T.; Hippo, Y.; Nakahori, Y.; Matsushita, I.; Kodama, T.; Nishimura, S.; Aburatani, H. Structure and chromosome location of human OGG1. Cytogenet. Genome Res. 1999, 85, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Dhénaut, A.; Boiteux, S.; Radicella, J.P. Characterization of the hOGG1 promoter and its expression during the cell cycle. Mutat. Res. 2000, 461, 109–118. [Google Scholar] [CrossRef]
- Arai, K.; Morishita, K.; Shinmura, K.; Kohno, T.; Kim, S.-R.; Nohmi, T.; Taniwaki, M.; Ohwada, S.; Yokota, J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene 1997, 14, 2857–2861. [Google Scholar] [CrossRef] [PubMed]
- Bjørås, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef]
- Kuo, F.C.; Sklar, J. Augmented expression of a human gene for 8-oxoguanine DNA glycosylase (MutM) in B lymphocytes of the dark zone in lymph node germinal centers. J. Exp. Med. 1997, 186, 1547–1556. [Google Scholar] [CrossRef]
- Lu, R.; Nash, H.M.; Verdine, G.L. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol. 1997, 7, 397–407. [Google Scholar] [CrossRef]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef]
- Roldán-Arjona, T.; Wei, Y.-F.; Carter, K.C.; Klungland, A.; Anselmino, C.; Wang, R.-P.; Augustus, M.; Lindahl, T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 8016–8020. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Shinmura, K.; Tosaka, M.; Tani, M.; Kim, S.-R.; Sugimura, H.; Nohmi, T.; Kasai, H.; Yokota, J. Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene 1998, 16, 3219–3225. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kohno, T.; Takeuchi-Sasaki, M.; Maeda, M.; Segawa, T.; Kamo, T.; Sugimura, H.; Yokota, J. Expression of the OGG1-type 1a (nuclear form) protein in cancerous and non-cancerous human cells. Int. J. Oncol. 2000, 16, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-H.; Yang, J.O.; Hahn, Y.; Kim, M.-R.; Byun, S.-S.; Jeon, Y.-J.; Kim, J.-M.; Song, K.-S.; Noh, S.-M.; Kim, S.; et al. Transcriptome analysis of human gastric cancer. Mamm. Genome 2005, 16, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Bruner, S.D.; Norman, D.P.G.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Lia, D.; Reyes, A.; de Melo Campos, J.T.A.; Piolot, T.; Baijer, J.; Radicella, J.P.; Campalans, A. Mitochondrial maintenance under oxidative stress depends on mitochondrially localised α-OGG1. J. Cell Sci. 2018, 131, jcs213538. [Google Scholar] [CrossRef]
- Audebert, M.; Charbonnier, J.B.; Boiteux, S.; Radicella, J.P. Mitochondrial targeting of human 8-oxoguanine DNA glycosylase hOGG1 is impaired by a somatic mutation found in kidney cancer. DNA Repair 2002, 1, 497–505. [Google Scholar] [CrossRef]
- Hashiguchi, K.; Stuart, J.A.; de Souza-Pinto, N.C.; Bohr, V.A. The C-terminal αO helix of human Ogg1 is essential for 8-oxoguanine DNA glycosylase activity: The mitochondrial β-Ogg1 lacks this domain and does not have glycosylase activity. Nucleic Acids Res. 2004, 32, 5596–5608. [Google Scholar] [CrossRef]
- Ogawa, A.; Watanabe, T.; Shoji, S.; Furihata, C. Enzyme kinetics of an alternative splicing isoform of mitochondrial 8-oxoguanine DNA glycosylase, OGG1-1b, and compared with the nuclear OGG1-1a. J. Biochem. Mol. Toxicol. 2015, 29, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; ap Rhys, C.M.J.; Hudson, E.K.; Dianov, G.L.; Hansford, R.G.; Bohr, V.A. An oxidative damage-specific endonuclease from rat liver mitochondria. J. Biol. Chem. 1997, 272, 27338–27344. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine DNA glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar] [PubMed]
- Matouschek, A.; Pfanner, N.; Voos, W. Protein unfolding by mitochondria: The Hsp70 import motor. EMBO Rep. 2000, 1, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Panduri, V.; Liu, G.; Surapureddi, S.; Kondapalli, J.; Soberanes, S.; de Souza-Pinto, N.C.; Bohr, V.A.; Budinger, G.R.S.; Schumacker, P.T.; Weitzman, S.A.; et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic. Biol. Med. 2009, 47, 750–759. [Google Scholar] [CrossRef]
- Su, Y.-H.; Lee, Y.-L.; Chen, S.-F.; Lee, Y.-P.; Hsieh, Y.-H.; Tsai, J.-H.; Hsu, J.-L.; Tian, W.-T.; Huang, W. Essential role of β-human 8-oxoguanine DNA glycosylase 1 in mitochondrial oxidative DNA repair. Environ. Mol. Mutagens. 2013, 54, 54–64. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Youn, H.-G.; Wang, H.-J.; Yoon, G. Decreased mitochondrial OGG1 expression is linked to mitochondrial defects and delayed hepatoma cell growth. Mol. Cells 2013, 35, 489–497. [Google Scholar] [CrossRef]
- Monden, Y.; Arai, T.; Asano, M.; Ohtsuka, E.; Aburatani, H.; Nishimura, S. Human MMH (OGG1) type 1a protein is a major enzyme for repair of 8-hydroxyguanine lesions in human cells. Biochem. Biophys. Res. Commun. 1999, 258, 605–610. [Google Scholar] [CrossRef]
- Hirano, T.; Kudo, H.; Doi, Y.; Nishino, T.; Fujimoto, S.; Tsurudome, Y.; Ootsuyama, Y.; Kasai, H. Detection of a smaller, 32-kDa 8-oxoguanine DNA glycosylase 1 in 3′-methyl-4-dimethylamino-azobenzene-treated mouse liver. Cancer Sci. 2004, 95, 118–122. [Google Scholar] [CrossRef]
- Hirano, T.; Kawai, K.; Ootsuyama, Y.; Orimo, H.; Kasai, H. Detection of a mouse OGG1 fragment during caspase-dependent apoptosis: Oxidative DNA damage and apoptosis. Cancer Sci. 2004, 95, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Fukae, J.; Takanashi, M.; Kubo, S.-I.; Nishioka, K.-I.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Kempisty, B.; Jaroszewska-Kolecka, J.; Rózycka, A.; Florczak, J.; Lianeri, M.; Jagodzinski, P.P.; Kozubski, W. Expression and polymorphisms of gene 8-oxoguanine glycosylase 1 and the level of oxidative DNA damage in peripheral blood lymphocytes of patients with Alzheimer’s disease. DNA Cell Biol. 2009, 28, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, F.; Viel, A.; Bignami, M. Role of MUTYH in human cancer. Mutat. Res. 2013, 743, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Banda, D.M.; Nuñez, N.N.; Burnside, M.A.; Bradshaw, K.M.; David, S.S. Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: Mechanism, metals and medicine. Free Radic. Biol. Med. 2017, 107, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Slupska, M.M.; Baikalov, C.; Luther, W.M.; Chiang, J.-H.; Wei, Y.-F.; Miller, J.H. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol. 1996, 178, 3885–3892. [Google Scholar] [CrossRef]
- Takao, M.; Zhang, Q.-M.; Yonei, S.; Yasui, A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine: 8-oxoguanine DNA glycosylase. Nucleic Acids Res. 1999, 27, 3638–3644. [Google Scholar] [CrossRef]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Shinmura, K.; Saitoh, T.; Takenoshita, S.; Kuwano, H.; Yokota, J. A single nucleotide polymorphism at the splice donor site of the human MYH base excision repair genes results in reduced translation efficiency of its transcripts. Genes Cells 2002, 7, 461–474. [Google Scholar] [CrossRef]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004, 32, 477–487. [Google Scholar] [CrossRef]
- De Belle, I.; Wu, J.-X.; Sperandio, S.; Mercola, D.; Adamson, E.D. In Vivo cloning and characterization of a new growth suppressor protein TOE1 as a direct target gene of Egr1. J. Biol. Chem. 2003, 278, 14306–14312. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Lee, H.M.; Englander, E.W. N-terminus of the rat adenine glycosylase MYH affects excision rates and processing of MYH-generated abasic sites. Nucleic Acids Res. 2004, 32, 4332–4339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.-H.; Singh, K.K.; Lu, A.-L. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001, 276, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Englander, E.W.; Hu, Z.; Sharma, A.; Lee, H.-M.; Wu, Z.-H.; Greeley, G.H. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J. Neurochem. 2002, 83, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Hu, Z.; Ma, H.; Greeley, G.H., Jr.; Wang, C.; Englander, E.W. Developmental changes in expression and subcellular localization of the DNA repair glycosylase, MYH, in the rat brain. J. Neurochem. 2004, 88, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C → T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Shinmura, K.; Hanaoka, T.; Natsukawa, S.; Shaura, K.; Koizumi, Y.; Kasuga, Y.; Ozawa, T.; Tsujinaka, T.; Li, Z.; et al. A novel splice-site variant of the base excision repair gene MYH is associated with production of an aberrant mRNA transcript encoding a truncated MYH protein not localized in the nucleus. Carcinogenesis 2004, 25, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Olschwang, S.; Blanché, H.; de Moncuit, C.; Thomas, G. Similar colorectal cancer risk in patients with monoallelic and biallelic mutations in the MYH gene identified in a population with adenomatous polyposis. Genet. Test. 2007, 11, 315–320. [Google Scholar] [CrossRef]
- Sampson, J.R.; Dolwani, S.; Jones, S.; Eccles, D.; Ellis, A.; Evans, D.G.; Frayling, I.; Jordan, S.; Maher, E.R.; Mak, T.; et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003, 362, 39–41. [Google Scholar] [CrossRef]
- Isidro, G.; Laranjeira, F.; Pires, A.; Leite, J.; Regateiro, F.; Castro e Sousa, F.; Soares, J.; Castro, C.; Giria, J.; Brito, M.J.; et al. Germline MUTYH (MYH) mutations in Portuguese individuals with multiple colorectal adenomas. Hum. Mutat. 2004, 24, 353–354. [Google Scholar] [CrossRef]
- Grandval, P.; Fabre, A.J.; Gaildrat, P.; Baert-Desurmont, S.; Blayau, M.; Buisine, M.P.; Coulet, F.; Maugard, C.; Pinson, S.; Remenieras, A.; et al. Genomic variations integrated database for MUTYH-associated adenomatous polyposis. J. Med. Genet. 2015, 52, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Uhlhaas, S.; Goergens, H.; Siberg, K.; Vogel, M.; Pagenstecher, C.; Mangold, E.; Caspari, R.; Propping, P.; Friedl, W. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int. J. Cancer. 2006, 119, 807–814. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Farwell, K.D.; Vuong, H.; Lu, H.M.; Mu, W.; Shahmirzadi, L.; Tang, S.; Chen, J.; Bhide, S.; Chao, E.C. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS ONE 2017, 12, e0170843. [Google Scholar] [CrossRef] [PubMed]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Hes, F.J.; Nagengast, F.M.; Weiss, M.M.; Mathus-Vliegen, E.M.; Morreau, H.; Breuning, M.H.; Wijnen, J.T.; Tops, C.M.; Vasen, H.F. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin. Genet. 2007, 71, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Out, A.A.; Tops, C.M.; Nielsen, M.; Weiss, M.M.; van Minderhout, I.J.; Fokkema, I.F.; Buisine, M.P.; Claes, K.; Colas, C.; Fodde, R.; et al. Leiden Open Variation Database of the MUTYH Gene. Hum. Mutat. 2010, 31, 1205–1215. [Google Scholar] [CrossRef]
- Jian, W.; Shao, K.; Qin, Q.; Wang, X.; Song, S.; Wang, X. Clinical and genetic characterization of hereditary breast cancer in a Chinese population. Hered. Cancer Clin. Pract. 2017, 15, 19. [Google Scholar] [CrossRef]
- Eliason, K.; Hendrickson, B.C.; Judkins, T.; Norton, M.; Leclair, B.; Lyon, E.; Ward, B.; Noll, W.; Scholl, T. The potential for increased clinical sensitivity in genetic testing for polyposis colorectal cancer through the analysis of MYH mutations in North American patients. J. Med. Genet. 2005, 42, 95–96. [Google Scholar] [CrossRef][Green Version]
- Yanaru-Fujisawa, R.; Matsumoto, T.; Ushijima, Y.; Esaki, M.; Hirahashi, M.; Gushima, M.; Yao, T.; Nakabeppu, Y.; Iida, M. Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin. Genet. 2008, 73, 545–553. [Google Scholar] [CrossRef]
- Ricci, M.T.; Miccoli, S.; Turchetti, D.; Bondavalli, D.; Viel, A.; Quaia, M.; Giacomini, E.; Gismondi, V.; Sanchez-Mete, L.; Stigliano, V.; et al. Type and frequency of MUTYH variants in Italian patients with suspected MAP: A retrospective multicenter study. J. Hum. Genet. 2017, 62, 309–315. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals with Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Choudhury, S.; Mitra, P.S.; Dubash, J.J.; Sajankila, S.P.; Roy, R. Targeting base excision repair for chemosensitization. Anticancer Agents Med. Chem. 2008, 8, 351–357. [Google Scholar] [CrossRef]
- O’Connor, T.R. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res. 1993, 21, 5561–5569. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.A.; Vyas, P.; Harris, P.C.; Simmons, D.L.; Higgs, D.R. Structure of the human 3-methyladenine DNA glycosylase gene and localization close to the 16p telomere. Proc. Natl. Acad. Sci. USA 1993, 90, 3437–3441. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Tatsuka, M.; Tano, K.; Asano, M.; Mitra, S. Molecular cloning and characterization of the promoter of the human N-methylpurine-DNA glycosylase (MPG) gene. Carcinogenesis 1997, 18, 1837–1839. [Google Scholar] [CrossRef][Green Version]
- Pendlebury, A.; Frayling, I.M.; Santibanez Koref, M.F.; Margison, G.P.; Rafferty, J.A. Evidence for the simultaneous expression of alternatively spliced alkylpurine N-glycosylase transcripts in human tissues and cells. Carcinogenesis 1994, 15, 2957–2960. [Google Scholar] [CrossRef]
- Bonanno, K.; Wyrzykowski, J.; Chong, W.; Matijasevic, Z.; Volkert, M.R. Alkylation resistance of E. coli cells expressing different isoforms of human alkyladenine DNA glycosylase (hAAG). DNA Repair 2002, 1, 507–516. [Google Scholar] [CrossRef]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. 2013, 743, 4–11. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, C.; Sengupta, S.; Mitra, S.; Hegde, M.L. New paradigms in the repair of oxidative damage in human genome: Mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell. Mol. Life Sci. 2015, 72, 1679–1698. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [PubMed]
- Takao, M.; Kanno, S.-I.; Kobayashi, K.; Zhang, Q.-M.; Yonei, S.; van der Horst, G.T.J.; Yasui, A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J. Biol. Chem. 2002, 277, 42205–42213. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Yamamoto, M.; Kusaka, H.; Masatsugu, H.; Matsuyama, S.; Tajima, T.; Ide, H.; Kubo, K. NEIL1 mRNA splicing variants are expressed in normal mouse organs. J. Radiat. Res. 2012, 53, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Matsuda, O.; Iba, K.; Terashima, I.; Sekiguchi, M.; Nakabeppu, Y. Molecular cloning of AtMMH, an Arabidopsis thaliana ortholog of the Escherichia coli mutM gene, and analysis of functional domains of its product. Mol. Gen. Genet. 1998, 259, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.-J.; Murphy, T.M. Alternative forms of formamidopyrimidine-DNA glycosylase from Arabidopsis thaliana. Photochem. Photobiol. 2001, 73, 128–134. [Google Scholar] [CrossRef]
- Murphy, T.M.; Gao, M.-J. Multiple forms of formamidopyrimidine-DNA glycosylase produced by alternative splicing in Arabidopsis thaliana. J. Photochem. Photobiol. B Biol. 2001, 61, 87–93. [Google Scholar] [CrossRef]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef] [PubMed]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjørås, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926–4936. [Google Scholar] [CrossRef]
- Demple, B.; Sung, J.-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Herman, T.; Chen, D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: Definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 11450–11454. [Google Scholar] [CrossRef] [PubMed]
- Robson, C.N.; Hickson, I.D. Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth (exonuclease III) mutants. Nucleic Acids Res. 1991, 19, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Bunville, J.; Patterson, T.A. Nucleotide sequence of a cDNA for an apurinic/apyrimidinic endonuclease from HeLa cells. Nucleic Acids Res. 1992, 20, 370. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Ascione, G.; Menninger, J.C.; Ward, D.C.; Demple, B. Human apurinic endonuclease gene (APE): Structure and genomic mapping (chromosome 1 4q11.2–12). Hum. Mol. Genet. 1992, 1, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Robson, C.N.; Hochhauser, D.; Craig, R.; Rack, K.; Bukie, V.J.; Hickson, I.D. Structure of the human DNA repair gene HAP1 and its localisation to chromosome 14q 11.2–12. Nucleic Acids Res. 1992, 20, 4417–4421. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Hatsushika, M.; Watanabe, S.; Akiyama, K.; Nagao, K.; Tsutsui, K. cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim. Biophys. Acta 1992, 1131, 287–299. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.-C.E.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Zhao, B.; Grandy, D.K.; Hagerup, J.M.; Magenis, R.E.; Smith, L.; Chauhan, B.C.; Hennee, W.D. The human gene fro apurinic/apyrimidinic endonuclease (HAP1): Sequence and localization to chromosome 14 band q12. Nucleic Acids Res. 1992, 20, 4097–4098. [Google Scholar] [CrossRef]
- Akiyama, K.; Seki, S.; Oshida, T.; Yoshida, M.C. Structure, promoter analysis and chromosomal assignment of the human APEX gene. Biochim. Biophys. Acta 1994, 1219, 15–25. [Google Scholar] [CrossRef]
- Yu, W.; Andersson, B.; Worley, K.C.; Muzny, D.M.; Ding, Y.; Liu, W.; Ricafrente, J.Y.; Wentland, M.A.; Lennon, G.; Gibbs, R.A. Large-scale concatenation cDNA sequencing. Genome Res. 1997, 7, 353–358. [Google Scholar] [CrossRef]
- Wang, A.-G.; Yoon, S.Y.; Oh, J.-H.; Jeon, Y.-J.; Kim, M.; Kim, J.-M.; Byun, S.-S.; Yang, J.O.; Kim, J.H.; Kim, D.-G.; et al. Identification of intrahepatic cholangiocarcinoma related genes by comparison with normal liver tissues using expressed sequence tags. Biochem. Biophys. Res. Commun. 2006, 345, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.W.; Horton, J.K.; Kühn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature 1996, 379, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Wilson, S.H. Structure and mechanism of DNA polymerase β. Chem. Rev. 2006, 106, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Çağlayan, M.; Dai, D.-P.; Nadalutti, C.A.; Zhao, M.-L.; Gassman, N.R.; Janoshazi, A.K.; Stefanick, D.F.; Horton, J.K.; Krasich, R.; et al. DNA polymerase β: A missing link of the base excision repair machinery in mammalian mitochondria. DNA Repair 2017, 60, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA polymerase beta participates in mitochondrial DNA repair. Mol. Cell. Biol. 2017, 37, e00237-17. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Beard, W.A.; Strauss, P.R.; Wilson, S.H. Human DNA polymerase β deoxyribose phosphate lyase: Substrate specificity and catalytic mechanism. J. Biol. Chem. 1998, 273, 15263–15270. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Prasad, R.; Wilson, S.H.; Bohr, V.A. Role of DNA polymerase β in the excision step of long patch mammalian base excision repair. J. Biol. Chem. 1999, 274, 13741–13743. [Google Scholar] [CrossRef]
- Podlutsky, A.J.; Dianova, I.I.; Podust, V.N.; Bohr, V.A.; Dianov, G.L. Human DNA polymerase β initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001, 20, 1477–1482. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Khodyreva, S.N.; Lebedeva, N.A.; Prasad, R.; Wilson, S.H.; Lavrik, O.I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease 1 (APE1), DNA polymerase β and poly(ADP-ribose) polymerase 1: Interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005, 33, 1222–1229. [Google Scholar] [CrossRef]
- Chyan, Y.-J.; Ackerman, S.; Shepherd, N.S.; McBride, O.W.; Widen, S.G.; Wilson, S.H.; Wood, T.G. The human DNA polymerase β gene structure. Evidence of alternative splicing in gene expression. Nucleic Acids Res. 1994, 22, 2719–2725. [Google Scholar] [CrossRef][Green Version]
- SenGupta, D.N.; Zmudzka, B.Z.; Kumar, P.; Cobianchi, F.; Skowronski, J.; Wilson, S.H. Sequence of human DNA polymerase β mRNA obtained through cDNA cloning. Biochem. Biophys. Res. Commun. 1986, 136, 341–347. [Google Scholar] [CrossRef]
- Zmudzka, B.Z.; SenGupta, D.; Matsukage, A.; Cobianchi, F.; Kumar, P.; Wilson, S.H. Structure of rat DNA polymerase β revealed by partial amino acid sequencing and cDNA cloning. Proc. Natl. Acad. Sci. USA 1986, 83, 5106–5110. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Beard, W.A.; Chyan, J.Y.; Maciejewski, M.W.; Mullen, G.P.; Wilson, S.H. Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase β as revealed by site-directed mutagenesis: DNA binding and 5′-deoxyribose phosphate lyase activities. J. Biol. Chem. 1998, 273, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Batra, V.K.; Yang, X.-P.; Krahn, J.M.; Pedersen, L.C.; Beard, W.A.; Wilson, S.H. Structural insight into the DNA polymerase β deoxyribose phosphate lyase mechanism. DNA Repair 2005, 4, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Zmudzka, B.Z.; Fomace, A.; Collins, J.; Wilson, S.H. Characterization of DNA polymerase β mRNA: Cell-cycle and growth response in cultured human cells. Nucleic Acids Res. 1988, 16, 9587–9596. [Google Scholar] [CrossRef]
- Chyan, Y.-J.; Strauss, P.R.; Wood, T.G.; Wilson, S.H. Identification of novel mRNA isoforms for human DNA polymerase β. DNA Cell. Biol. 1996, 15, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.; Chen, H.-C.; Comhair, S.; Erzurum, S.C.; Banerjee, S. Variant forms of DNA polymerase β in primary lung carcinomas. DNA Cell. Biol. 1999, 18, 549–554. [Google Scholar] [CrossRef]
- Thompson, T.E.; Rogan, P.K.; Risinger, J.I.; Taylor, J.A. Splice variants but not mutations of DNA polymerase β are common in bladder cancer. Cancer Res. 2002, 62, 3251–3256. [Google Scholar]
- Skandalis, A.; Uribe, E. A survey of splice variants of the human hypoxanthine phosphoribosyl transferase and DNA polymerase beta genes: Products of alternative or aberrant splicing? Nucleic Acids Res. 2004, 32, 6557–6564. [Google Scholar] [CrossRef][Green Version]
- Disher, K.; Skandalis, A. Evidence of the modulation of mRNA splicing fidelity in humans by oxidative stress and p53. Genome 2007, 50, 946–953. [Google Scholar] [CrossRef]
- Simonelli, V.; D’Errico, M.; Palli, D.; Prasad, R.; Wilson, S.H.; Dogliotti, E. Characterization of DNA polymerase β splicing variants in gastric cancer: The most frequent exon 2-deleted isoform is a non-coding RNA. Mutat. Res. 2009, 670, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, A.; Frampton, M.; Seger, J.; Richards, M.H. The adaptive significance of unproductive alternative splicing in primates. RNA 2010, 16, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, A. Estimation of the minimum mRNA splicing error rate in vertebrates. Mutat. Res. 2016, 784, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.; Chen, H.-C.; Grundfest-Broniatowski, S.; Banerjee, S. Alteration of hMSH2 and DNA polymerase β genes in breast carcinomas and fibroadenomas. Biochem. Biophys. Res. Commun. 1999, 259, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Bhattacharyya, N.; Wang, L.; Recupero, A.J.; Klein, E.A.; Harter, M.L.; Banerjee, S. Defective DNA repair genes in a primary culture of human renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2000, 126, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Zheng, N.G.; Wu, J.L.; Li, S.K.; Wang, Y.L. Difference in expression level and localization of DNA polymerase beta among human esophageal cancer focus, adjacent and corresponding normal tissues. Dis. Esophagus 2006, 19, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Patel, U.; Ghosh, L.; Banerjee, S. DNA polymerase β mutations in human colorectal cancer. Cancer Res. 1992, 52, 4824–4827. [Google Scholar]
- Sadakane, Y.; Maeda, K.; Kuroda, Y.; Hori, K. Identification of mutations in DNA polymerase β mRNAs from patients with Werner syndrome. Biochem. Biophys. Res. Commun. 1994, 200, 219–225. [Google Scholar] [CrossRef]
- Wang, L.; Banerjee, S. Mutations in DNA polymerase β occur in breast, prostate and colorectal tumors. Int. J. Oncol. 1995, 6, 459–463. [Google Scholar] [CrossRef]
- Bhattacharyya, N.; Banerjee, S. A variant of DNA polymerase β acts as a dominant negative mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 10324–10329. [Google Scholar] [CrossRef]
- Bhattacharyya, N.; Banerjee, T.; Patel, U.; Banerjee, S. Impaired repair activity of a truncated DNA polymerase β protein. Life Sci. 2001, 69, 271–280. [Google Scholar] [CrossRef]
- Wang, L.; Bhattacharyya, N.; Rabi, T.; Wang, L.; Banerjee, S. Mammary carcinogenesis in transgenic mice expressing a dominant-negative mutant of DNA polymerase β in their mammary glands. Carcinogenesis 2007, 28, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.; Banerjee, S. A novel role of XRCC1 in the functions of a DNA polymerase β variant. Biochemistry 2001, 40, 9005–9013. [Google Scholar] [CrossRef] [PubMed]
- Khanra, K.; Bhattacharya, C.; Bhattacharyya, N. Association of a newly identified variant of DNA polymerase beta (polβΔ63-123, 208-304) with the risk factor of ovarian carcinoma in India. Asian Pac. J. Cancer Prev. 2012, 13, 1999–2002. [Google Scholar] [CrossRef]
- Khanra, K.; Chakraborty, A.; Bhattacharyya, N. HeLa cells containing a truncated form of DNA polymerase beta are more sensitized to alkylating agents than to agents inducing oxidative stress. Asian Pac. J. Cancer Prev. 2015, 16, 8177–8186. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.; Chen, H.-C.; Wang, L.; Banerjee, S. Heterogeneity in expression of DNA polymerase β and DNA repair activity in human tumor cell lines. Gene Expr. 2002, 10, 115–123. [Google Scholar] [CrossRef]
- Casas-Finet, J.R.; Kumar, A.; Karpel, R.L.; Wilson, S.H. Mammalian DNA polymerase β: Characterization of a 16-kDa transdomain fragment containing the nucleic acid-binding activities of the native enzyme. Biochemistry 1992, 31, 10272–10280. [Google Scholar] [CrossRef] [PubMed]
- Nowak, R.; Bieganowski, P.; Konopiński, R.; Siedlecki, J.A. Alternative splicing of DNA polymerase β mRNA is not tumor-specific. Int. J. Cancer 1996, 68, 199–202. [Google Scholar] [CrossRef]
- Grosso, A.R.; Martins, S.; Carmo-Fonseca, M. The emerging role of splicing factors in cancer. EMBO Rep. 2008, 9, 1087–1093. [Google Scholar] [CrossRef]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).