Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities



Abstract
1. Introduction
2. Alloimmune Response
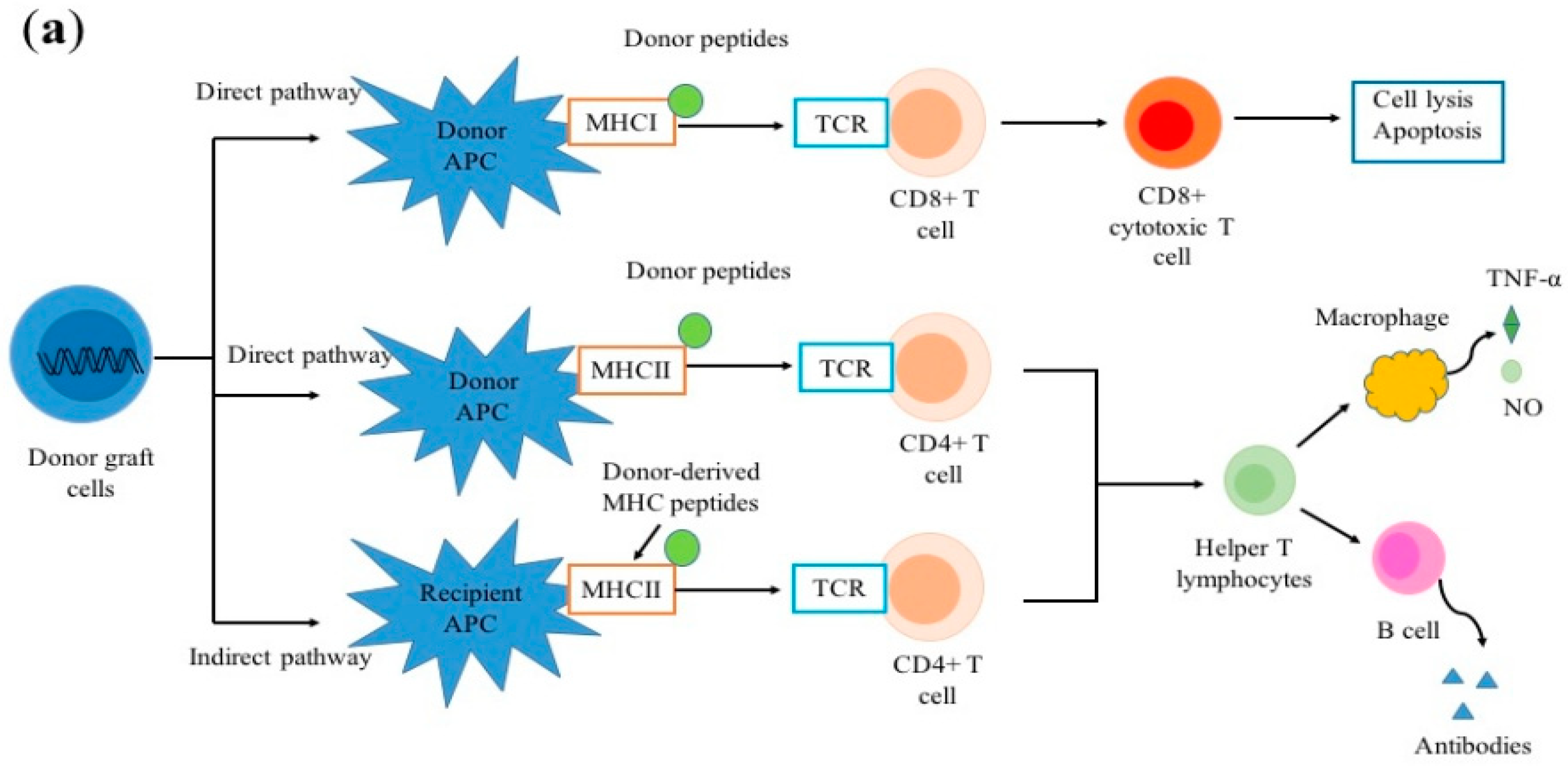
2.1. Allorecognition
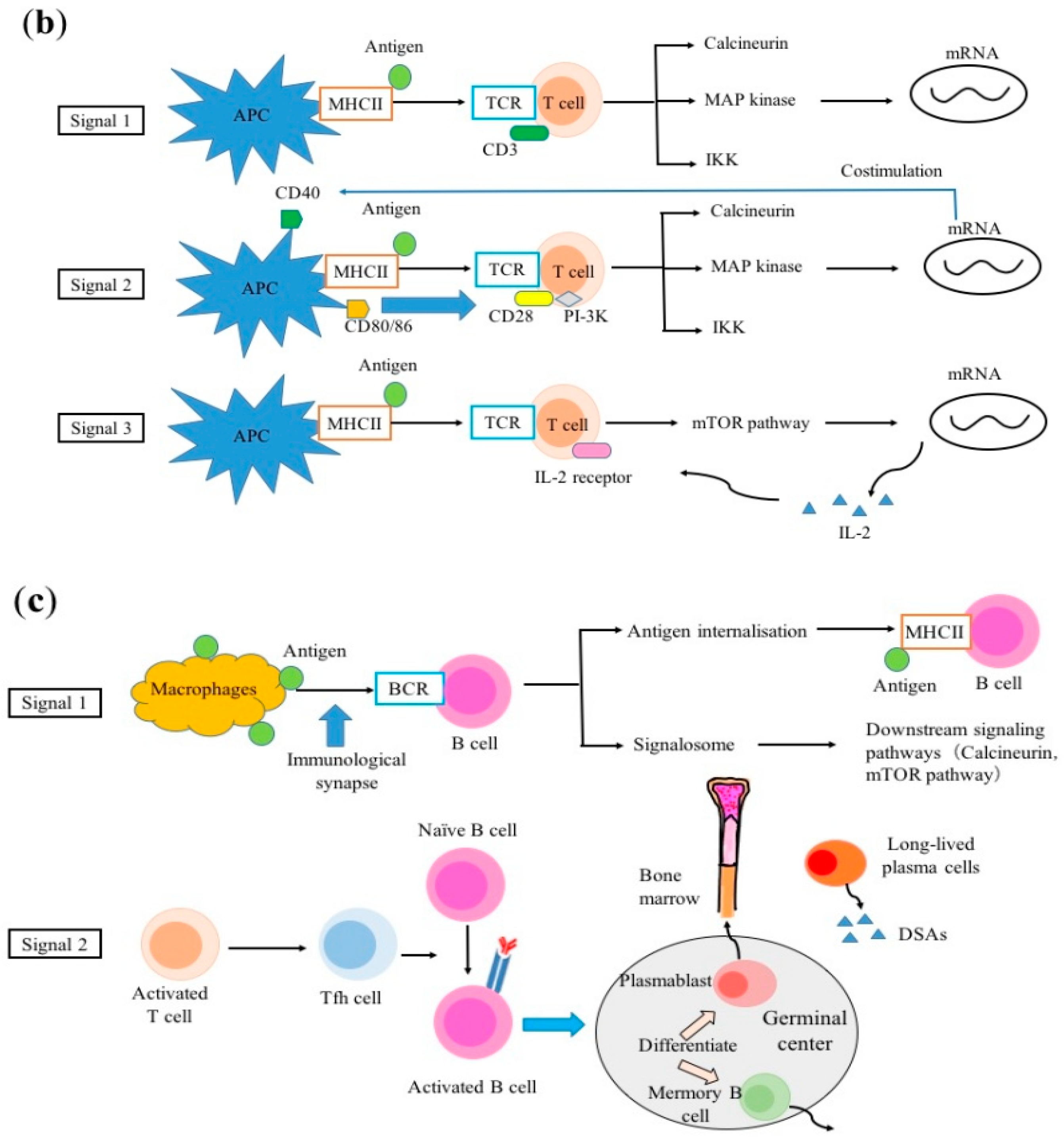
2.2. T-cell Activation
2.3. B-cell Activation
3. Classification of Immunosuppressants
4. Clinical Features Induced by Different Immunosuppressants
4.1. CNIs
4.2. Antimetabolites
4.3. Corticosteroids
4.4. Monoclonal Antibodies
5. Mechanisms of Neurotoxicity Induced by Different Immunosuppressants
5.1. CNIs
5.2. Antimetabolites
5.3. Corticosteroids
5.4. Monoclonal Antibodies
6. Management
7. Neuroprotective Effects
7.1. CNIs
7.2. mTOR Inhibitors
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB1 | ATP-binding cassette transporter B1 |
| ADC | Apparent diffusion coefficient |
| APC | Antigen presenting cell |
| ATP | Adenosine triphosphate |
| BBB | Blood-brain barrier |
| BCR | B-cell receptor |
| BDNF | Brain-derived neurotrophic factor |
| CD | Cluster of differentiation |
| CNI | Calcineurin inhibitor |
| CNS | Central nervous system |
| CPM | Central pontine myelinolysis |
| CS | Citrate synthase |
| CsA | Cyclosporin A |
| CT | Computed tomography |
| CYP | Cytochrome pigment |
| DWI | Diffusion weighted image |
| EC | Endothelial cell |
| EEG | Electro-encephalogram |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| fEPSP | Field excitatory postsynaptic potentials |
| FKBP | FK506-binding protein |
| FLAIR | Fluid-attenuated IR |
| IFN-γ | Interferon-gamma |
| IL-2 | Interleukin-2 |
| IMPDH | Inosine-5′-monophosphate dehydrogenase |
| IP3 | Inositol trisphosphate |
| JAK | Janus kinase |
| MAP | Mitogen activated protein |
| MBEC4 | Mouse brain capillary endothelial cells |
| MHC | Major histocompatibility complex |
| mPT | Mitochondrial permeability transition |
| MRI | Magnetic resonance imaging |
| mTOR | Mammalian target of rapamycin |
| MTX | Methotrexate |
| muromonab-CD3 | Mouse monoclonal immunoglobulin G2 antibody to cluster of differentiation 3 |
| NFAT | Nuclear factor of activated T cells |
| NFκB | Nuclear factor kappa B |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| P-gp | P-glycoprotein |
| PRES | Posterior reversible encephalopathy syndrome |
| PTEN | Phosphatase and tensin homolog |
| RAS | Renin–angiotensin system |
| RC | Respiratory chain |
| ROS | Reactive oxygen species |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| S6K1 | S6 kinase 1 |
| TCR | T-cell receptor |
| Tfh | T follicular helper |
| TNF-α | Tumor necrosis factor-alpha |
| TrkA | Tropomyosin receptor kinase A |
| TrkB | Tyrosine kinase receptor B |
References
- Murray, J.E.; Merrill, J.P.; Harrison, J.H. Renal homotransplantation in identical twins. 1955. J. Am. Soc. Nephrol. 2001, 12, 201–204. [Google Scholar] [PubMed]
- Tolou-Ghamari, Z. Nephro and neurotoxicity of calcineurin inhibitors and mechanisms of rejections: A review on tacrolimus and cyclosporin in organ transplantation. J. Nephropathol. 2012, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Van Sandwijk, M.S.; Bemelman, F.J.; Ten Berge, I.J. Immunosuppressive drugs after solid organ transplantation. Neth. J. Med. 2013, 71, 281–289. [Google Scholar] [PubMed]
- Coelho, T.; Tredger, M.; Dhawan, A. Current status of immunosuppressive agents for solid organ transplantation in children. Pediatr. Transplant. 2012, 16, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Mazariegos, G.V.; Molmenti, E.P.; Kramer, D.J. Early complications after orthotopic liver transplantation. Surg. Clin. N. Am. 1999, 79, 109–129. [Google Scholar] [CrossRef]
- Pruitt, A.A. Neurologic Complications of Transplantation. Continuum. (Minneap Minn) 2017, 23, 802–821. [Google Scholar] [CrossRef]
- Pizzi, M.; Ng, L. Neurologic Complications of Solid Organ Transplantation. Neurol. Clin. 2017, 35, 809–823. [Google Scholar] [CrossRef]
- Van de Beek, D.; Kremers, W.; Daly, R.C.; Edwards, B.S.; Clavell, A.L.; McGregor, C.G.; Wijdicks, E.F. Effect of neurologic complications on outcome after heart transplant. Arch. Neurol. 2008, 65, 226–231. [Google Scholar] [CrossRef][Green Version]
- Ardizzone, G.; Arrigo, A.; Schellino, M.M.; Stratta, C.; Valzan, S.; Skurzak, S.; Andruetto, P.; Panio, A.; Ballaris, M.A.; Lavezzo, B.; et al. Neurological complications of liver cirrhosis and orthotopic liver transplant. Transplant. Proc. 2006, 38, 789–792. [Google Scholar] [CrossRef]
- Ponticelli, C.; Campise, M.R. Neurological complications in kidney transplant recipients. J. Nephrol. 2005, 18, 521–528. [Google Scholar]
- Ocal, R.; Kibaroglu, S.; Derle, E.; Tanoglu, C.; Camkiran, A.; Pirat, A.; Can, U.; Sezgin, A. Neurologic Complications After Cardiac Transplant. Exp. Clin. Transplant. 2016. [Google Scholar] [CrossRef][Green Version]
- Dowling, M.R.; Li, S.; Dey, B.R.; McAfee, S.L.; Hock, H.R.; Spitzer, T.R.; Chen, Y.B.; Ballen, K.K. Neurologic complications after allogeneic hematopoietic stem cell transplantation: Risk factors and impact. Bone Marrow. Transplant. 2018, 53, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Wijdicks, E.F. Neurotoxicity of immunosuppressive drugs. Liver Transpl. 2001, 7, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Bechstein, W.O. Neurotoxicity of calcineurin inhibitors: Impact and clinical management. Transpl. Int. 2000, 13, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Dhar, R.; Human, T. Central nervous system complications after transplantation. Neurol. Clin. 2011, 29, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Boardman, D.A.; Jacob, J.; Smyth, L.A.; Lombardi, G.; Lechler, R.I. What Is Direct Allorecognition? Curr. Transplant. Rep. 2016, 3, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, M.R.; Lombardi, G.; Lechler, R.I. The importance of the indirect pathway of allorecognition in clinical transplantation. Curr. Opin. Immunol. 2008, 20, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.A.; Kohli, J.; Bloom, R.D. Immunosuppression for kidney transplantation: Where are we now and where are we going? Transplant. Rev. (Orlando) 2017, 31, 10–17. [Google Scholar] [CrossRef]
- Thaunat, O.; Granja, A.G.; Barral, P.; Filby, A.; Montaner, B.; Collinson, L.; Martinez-Martin, N.; Harwood, N.E.; Bruckbauer, A.; Batista, F.D. Asymmetric segregation of polarized antigen on B cell division shapes presentation capacity. Science 2012, 335, 475–479. [Google Scholar] [CrossRef]
- Harwood, N.E.; Batista, F.D. Early events in B cell activation. Annu. Rev. Immunol. 2010, 28, 185–210. [Google Scholar] [CrossRef]
- Schnyder, T.; Castello, A.; Feest, C.; Harwood, N.E.; Oellerich, T.; Urlaub, H.; Engelke, M.; Wienands, J.; Bruckbauer, A.; Batista, F.D. B cell receptor-mediated antigen gathering requires ubiquitin ligase Cbl and adaptors Grb2 and Dok-3 to recruit dynein to the signaling microcluster. Immunity 2011, 34, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Koenig, A.; Saison, C.; Dahdal, S.; Rigault, G.; Barba, T.; Taillardet, M.; Chartoire, D.; Ovize, M.; Morelon, E.; et al. CD4+ T Cell Help Is Mandatory for Naive and Memory Donor-Specific Antibody Responses: Impact of Therapeutic Immunosuppression. Front. Immunol. 2018, 9, 275. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Miller, M.J.; Parker, I.; Krummel, M.F.; Neighbors, M.; Hartley, S.B.; O’Garra, A.; Cahalan, M.D.; Cyster, J.G. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 2005, 3, e150. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D. SnapShot: The germinal center reaction. Cell 2014, 159, 700–700.e701. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Phares, T.W.; Yu, H.; Fan, R.; Baldwin, W.M., 3rd; Fairchild, R.L.; Valujskikh, A. The spleen is the major source of antidonor antibody-secreting cells in murine heart allograft recipients. Am. J. Transplant. 2012, 12, 1708–1719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, A.L.; Watson, C.J.; Bradley, J.A. Immunosuppressive agents in solid organ transplantation: Mechanisms of action and therapeutic efficacy. Crit. Rev. Oncol. Hematol. 2005, 56, 23–46. [Google Scholar] [CrossRef]
- Shin, H.S.; Grgic, I.; Chandraker, A. Novel Targets of Immunosuppression in Transplantation. Clin. Lab. Med. 2019, 39, 157–169. [Google Scholar] [CrossRef]
- Holt, C.D. Overview of Immunosuppressive Therapy in Solid Organ Transplantation. Anesthesiol. Clin. 2017, 35, 365–380. [Google Scholar] [CrossRef]
- McDermott, J.K.; Girgis, R.E. Individualizing immunosuppression in lung transplantation. Glob. Cardiol. Sci. Pract. 2018, 2018, 5. [Google Scholar] [CrossRef][Green Version]
- Thaunat, O.; Koenig, A.; Leibler, C.; Grimbert, P. Effect of Immunosuppressive Drugs on Humoral Allosensitization after Kidney Transplant. J. Am. Soc. Nephrol. 2016, 27, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Furiasse, N.; Kobashigawa, J.A. Immunosuppression and adult heart transplantation: Emerging therapies and opportunities. Expert Rev. Cardiovasc. Ther. 2017, 15, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Shapiro, R. New immunosuppressive agents in pediatric transplantation. Clinics (Sao Paulo) 2014, 69 (Suppl. 1), 8–16. [Google Scholar] [CrossRef]
- Mueller, A.R.; Platz, K.P.; Bechstein, W.O.; Schattenfroh, N.; Stoltenburg-Didinger, G.; Blumhardt, G.; Christe, W.; Neuhaus, P. Neurotoxicity after orthotopic liver transplantation. A comparison between cyclosporine and FK506. Transplantation 1994, 58, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Bartynski, W.S.; Zeigler, Z.R.; Shadduck, R.K.; Lister, J. Pretransplantation conditioning influence on the occurrence of cyclosporine or FK-506 neurotoxicity in allogeneic bone marrow transplantation. AJNR Am. J. Neuroradiol. 2004, 25, 261–269. [Google Scholar] [PubMed]
- Campagna, F.; Biancardi, A.; Cillo, U.; Gatta, A.; Amodio, P. Neurocognitive-neurological complications of liver transplantation: A review. Metab. Brain Dis. 2010, 25, 115–124. [Google Scholar] [CrossRef]
- Fujii, N.; Ikeda, K.; Koyama, M.; Aoyama, K.; Masunari, T.; Kondo, E.; Matsuzaki, T.; Mizobuchi, S.; Hiraki, A.; Teshima, T.; et al. Calcineurin inhibitor-induced irreversible neuropathic pain after allogeneic hematopoietic stem cell transplantation. Int. J. Hematol. 2006, 83, 459–461. [Google Scholar] [CrossRef]
- Gmitterova, K.; Minar, M.; Zigrai, M.; Kosutzka, Z.; Kusnirova, A.; Valkovic, P. Tacrolimus-induced parkinsonism in a patient after liver transplantation - case report. BMC Neurol. 2018, 18, 44. [Google Scholar] [CrossRef]
- Chopra, A.; Das, P.; Rai, A.; Kuppuswamy, P.S.; Li, X.; Huston, J.; Philbrick, K.; Sola, C. Catatonia as a manifestation of tacrolimus-induced neurotoxicity in organ transplant patients: A case series. Gen. Hosp. Psychiatry 2012, 34, e209–e211. [Google Scholar] [CrossRef]
- Scheel, A.K.; Blaschke, S.; Schettler, V.; Mayer, C.; Muller, G.A.; Bittermann, H.J.; Grunewald, R.W. Severe neurotoxicity of tacrolimus (FK506) after renal transplantation: Two case reports. Transplant. Proc. 2001, 33, 3693–3694. [Google Scholar] [CrossRef]
- Erro, R.; Bacchin, R.; Magrinelli, F.; Tomei, P.; Geroin, C.; Squintani, G.; Lupo, A.; Zaza, G.; Tinazzi, M. Tremor induced by Calcineurin inhibitor immunosuppression: A single-centre observational study in kidney transplanted patients. J. Neurol. 2018, 265, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, S.A.; Abdel-Hamid, H. Neurologic manifestations of transplant complications. Neurol. Clin. 2010, 28, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D., Jr.; Adler, B.; Turner, T.L.; Mansour, H.M. Alternative tacrolimus and sirolimus regimen associated with rapid resolution of posterior reversible encephalopathy syndrome after lung transplantation. Pediatr. Neurol. 2014, 50, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Kiemeneij, I.M.; de Leeuw, F.E.; Ramos, L.M.; van Gijn, J. Acute headache as a presenting symptom of tacrolimus encephalopathy. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1126–1127. [Google Scholar] [CrossRef] [PubMed]
- Steg, R.E.; Kessinger, A.; Wszolek, Z.K. Cortical blindness and seizures in a patient receiving FK506 after bone marrow transplantation. Bone Marrow Transplant. 1999, 23, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Hodnett, P.; Coyle, J.; O’Regan, K.; Maher, M.M.; Fanning, N. PRES (posterior reversible encephalopathy syndrome), a rare complication of tacrolimus therapy. Emerg. Radiol. 2009, 16, 493–496. [Google Scholar] [CrossRef]
- Lee, V.H.; Wijdicks, E.F.; Manno, E.M.; Rabinstein, A.A. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch. Neurol. 2008, 65, 205–210. [Google Scholar] [CrossRef]
- Wu, Q.; Marescaux, C.; Wolff, V.; Jeung, M.Y.; Kessler, R.; Lauer, V.; Chen, Y. Tacrolimus-associated posterior reversible encephalopathy syndrome after solid organ transplantation. Eur. Neurol. 2010, 64, 169–177. [Google Scholar] [CrossRef]
- Bartynski, W.S.; Tan, H.P.; Boardman, J.F.; Shapiro, R.; Marsh, J.W. Posterior reversible encephalopathy syndrome after solid organ transplantation. AJNR Am. J. Neuroradiol. 2008, 29, 924–930. [Google Scholar] [CrossRef]
- Cruz, R.J., Jr.; DiMartini, A.; Akhavanheidari, M.; Iacovoni, N.; Boardman, J.F.; Donaldson, J.; Humar, A.; Bartynski, W.S. Posterior reversible encephalopathy syndrome in liver transplant patients: Clinical presentation, risk factors and initial management. Am. J. Transplant. 2012, 12, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; David, V.G.; Varughese, S.; Tamilarasi, V.; Jacob, C.K. Posterior reversible encephalopathy syndrome in a renal allograft recipient: A complication of immunosuppression? Indian J. Nephrol. 2013, 23, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bonham, A.; Fukui, M. Immunosuppressive-associated leukoencephalopathy in organ transplant recipients. Transplantation 2000, 69, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, D.J.; Luetmer, P.H.; Campeau, N.G. Posterior reversible encephalopathy syndrome: Prognostic utility of quantitative diffusion-weighted MR images. AJNR Am. J. Neuroradiol. 2002, 23, 1038–1048. [Google Scholar] [PubMed]
- Hinchey, J.; Chaves, C.; Appignani, B.; Breen, J.; Pao, L.; Wang, A.; Pessin, M.S.; Lamy, C.; Mas, J.L.; Caplan, L.R. A reversible posterior leukoencephalopathy syndrome. N. Engl. J. Med. 1996, 334, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Fattapposta, F.; Locuratolo, N.; Pierallini, A.; Rossi, M.; Ruberto, F.; Bozzao, L. Reversible diffusion MRI abnormalities and transient mutism after liver transplantation. Neurology 2004, 62, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Bartynski, W.S.; Zeigler, Z.; Spearman, M.P.; Lin, L.; Shadduck, R.K.; Lister, J. Etiology of cortical and white matter lesions in cyclosporin-A and FK-506 neurotoxicity. AJNR Am. J. Neuroradiol. 2001, 22, 1901–1914. [Google Scholar]
- Kumar, S.; Fowler, M.; Gonzalez-Toledo, E.; Jaffe, S.L. Central pontine myelinolysis, an update. Neurol. Res. 2006, 28, 360–366. [Google Scholar] [CrossRef]
- Lampl, C.; Yazdi, K. Central pontine myelinolysis. Eur. Neurol. 2002, 47, 3–10. [Google Scholar] [CrossRef]
- De Groen, P.C.; Aksamit, A.J.; Rakela, J.; Forbes, G.S.; Krom, R.A. Central nervous system toxicity after liver transplantation. The role of cyclosporine and cholesterol. N. Engl. J. Med. 1987, 317, 861–866. [Google Scholar] [CrossRef]
- Truwit, C.L.; Denaro, C.P.; Lake, J.R.; DeMarco, T. MR imaging of reversible cyclosporin A-induced neurotoxicity. AJNR Am. J. Neuroradiol. 1991, 12, 651–659. [Google Scholar] [PubMed]
- Kusztal, M.; Piotrowski, P.; Mazanowska, O.; Misiak, B.; Kantorska-Janiec, M.; Boratynska, M.; Klinger, M.; Kiejna, A. Catatonic episode after kidney transplantation. Gen. Hosp. Psychiatry 2014, 36, e363–e365. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Hidalgo, F.; Martinez-Salio, A.; Moreno-Garcia, S.; de Pablo-Fernandez, E.; Correas-Callero, E.; Ruiz-Morales, J. Akinetic mutism induced by tacrolimus. Clin. Neuropharmacol. 2009, 32, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Di Nuzzo, S.; Zanni, M.; De Panfilis, G. Exacerbation of paranoid schizophrenia in a psoriatic patient after treatment with cyclosporine A, but not with etanercept. J. Drugs Dermatol. 2007, 6, 1046–1047. [Google Scholar] [PubMed]
- Draper, H.M. Depressive disorder associated with mycophenolate mofetil. Pharmacotherapy 2008, 28, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.S. Effects of glucocorticoids on mood, memory, and the hippocampus. Treatment and preventive therapy. Ann. N. Y. Acad. Sci. 2009, 1179, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Tzachanis, D.; Haider, M.; Papazisis, G. A Case of Subacute Encephalopathy Developing After Treatment With Clofarabine and Methotrexate That Resolved With Corticosteroids. Am. J. Ther. 2016, 23, e937–e940. [Google Scholar] [CrossRef] [PubMed]
- Vezmar, S.; Becker, A.; Bode, U.; Jaehde, U. Biochemical and clinical aspects of methotrexate neurotoxicity. Chemotherapy 2003, 49, 92–104. [Google Scholar] [CrossRef]
- Amodio, P.; Biancardi, A.; Montagnese, S.; Angeli, P.; Iannizzi, P.; Cillo, U.; D’Amico, D.; Gatta, A. Neurological complications after orthotopic liver transplantation. Dig. Liver Dis. 2007, 39, 740–747. [Google Scholar] [CrossRef]
- Hochberg, F.H.; Miller, D.C. Primary central nervous system lymphoma. J. Neurosurg. 1988, 68, 835–853. [Google Scholar] [CrossRef]
- Wolkowitz, O.M.; Lupien, S.J.; Bigler, E.; Levin, R.B.; Canick, J. The “steroid dementia syndrome”: An unrecognized complication of glucocorticoid treatment. Ann. N. Y. Acad. Sci. 2004, 1032, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Anghel, D.; Tanasescu, R.; Campeanu, A.; Lupescu, I.; Podda, G.; Bajenaru, O. Neurotoxicity of immunosuppressive therapies in organ transplantation. Maedica (Buchar) 2013, 8, 170–175. [Google Scholar] [PubMed]
- Fessler, R.G.; Johnson, D.L.; Brown, F.D.; Erickson, R.K.; Reid, S.A.; Kranzler, L. Epidural lipomatosis in steroid-treated patients. Spine (Phila Pa 1976) 1992, 17, 183–188. [Google Scholar] [CrossRef]
- Richards, J.M.; Vogelzang, N.J.; Bluestone, J.A. Neurotoxicity after treatment with muromonab-CD3. N. Engl. J. Med. 1990, 323, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Thaisetthawatkul, P.; Weinstock, A.; Kerr, S.L.; Cohen, M.E. Muromonab-CD3-induced neurotoxicity: Report of two siblings, one of whom had subsequent cyclosporin-induced neurotoxicity. J. Child. Neurol. 2001, 16, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Rabinstein, A.A.; Edwards, B.S.; Wijdicks, E.F. OKT3 neurotoxicity presenting as akinetic mutism. Transplantation 2003, 75, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.J.; Benani, D.J. A review of blinatumomab, a novel immunotherapy. J. Oncol. Pharm. Pract. 2016, 22, 639–645. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, T.W.; Feng, A.C.; Fan, H.L.; Hsieh, C.B.; Chung, K.P. Comprehensive risk assessment for early neurologic complications after liver transplantation. World J. Gastroenterol. 2016, 22, 5548–5557. [Google Scholar] [CrossRef]
- Erer, B.; Polchi, P.; Lucarelli, G.; Angelucci, E.; Baronciani, D.; Galimberti, M.; Giardini, C.; Gaziev, D.; Maiello, A. CsA-associated neurotoxicity and ineffective prophylaxis with clonazepam in patients transplanted for thalassemia major: Analysis of risk factors. Bone Marrow. Transplant. 1996, 18, 157–162. [Google Scholar]
- Craven, J.L. Cyclosporine-associated organic mental disorders in liver transplant recipients. Psychosomatics 1991, 32, 94–102. [Google Scholar] [CrossRef]
- Yanagimachi, M.; Naruto, T.; Tanoshima, R.; Kato, H.; Yokosuka, T.; Kajiwara, R.; Fujii, H.; Tanaka, F.; Goto, H.; Yagihashi, T.; et al. Influence of CYP3A5 and ABCB1 gene polymorphisms on calcineurin inhibitor-related neurotoxicity after hematopoietic stem cell transplantation. Clin. Transplant. 2010, 24, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Ieiri, I.; Kataoka, Y.; Tanabe, M.; Nishizaki, T.; Oishi, R.; Higuchi, S.; Otsubo, K.; Sugimachi, K. Neurotoxicity induced by tacrolimus after liver transplantation: Relation to genetic polymorphisms of the ABCB1 (MDR1) gene. Transplantation 2002, 74, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.K.; Senior, P.A. Tacrolimus toxicity in islet transplantation due to interaction with macrolides. Clin. Diabetes Endocrinol. 2016, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Balderramo, D.; Prieto, J.; Cardenas, A.; Navasa, M. Hepatic encephalopathy and post-transplant hyponatremia predict early calcineurin inhibitor-induced neurotoxicity after liver transplantation. Transpl. Int. 2011, 24, 812–819. [Google Scholar] [CrossRef] [PubMed]
- DiMartini, A.; Fontes, P.; Dew, M.A.; Lotrich, F.E.; de Vera, M. Age, model for end-stage liver disease score, and organ functioning predict posttransplant tacrolimus neurotoxicity. Liver Transpl. 2008, 14, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Bottiger, Y.; Brattstrom, C.; Tyden, G.; Sawe, J.; Groth, C.G. Tacrolimus whole blood concentrations correlate closely to side-effects in renal transplant recipients. Br. J. Clin. Pharmacol. 1999, 48, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Lauerma, A.I.; Surber, C.; Maibach, H.I. Absorption of topical tacrolimus (FK506) in vitro through human skin: Comparison with cyclosporin A. Skin Pharmacol. 1997, 10, 230–234. [Google Scholar] [CrossRef]
- Tanaka, K.; Hirai, M.; Tanigawara, Y.; Yasuhara, M.; Hori, R.; Ueda, K.; Inui, K. Effect of cyclosporin analogues and FK506 on transcellular transport of daunorubicin and vinblastine via P-glycoprotein. Pharm. Res. 1996, 13, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Kochi, S.; Takanaga, H.; Matsuo, H.; Ohtani, H.; Naito, M.; Tsuruo, T.; Sawada, Y. Induction of apoptosis in mouse brain capillary endothelial cells by cyclosporin A and tacrolimus. Life Sci. 2000, 66, 2255–2260. [Google Scholar] [CrossRef]
- Dohgu, S.; Yamauchi, A.; Nakagawa, S.; Takata, F.; Kai, M.; Egawa, T.; Naito, M.; Tsuruo, T.; Sawada, Y.; Niwa, M.; et al. Nitric oxide mediates cyclosporine-induced impairment of the blood-brain barrier in cocultures of mouse brain endothelial cells and rat astrocytes. Eur. J. Pharmacol. 2004, 505, 51–59. [Google Scholar] [CrossRef]
- Kochi, S.; Takanaga, H.; Matsuo, H.; Naito, M.; Tsuruo, T.; Sawada, Y. Effect of cyclosporin A or tacrolimus on the function of blood-brain barrier cells. Eur. J. Pharmacol. 1999, 372, 287–295. [Google Scholar] [CrossRef]
- Fabulas-da Costa, A.; Aijjou, R.; Hachani, J.; Landry, C.; Cecchelli, R.; Culot, M. In vitro blood-brain barrier model adapted to repeated-dose toxicological screening. Toxicol. In Vitro 2013, 27, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Bellwon, P.; Culot, M.; Wilmes, A.; Schmidt, T.; Zurich, M.G.; Schultz, L.; Schmal, O.; Gramowski-Voss, A.; Weiss, D.G.; Jennings, P.; et al. Cyclosporine A kinetics in brain cell cultures and its potential of crossing the blood-brain barrier. Toxicol. In Vitro 2015, 30, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.; Zurich, M.G.; Culot, M.; da Costa, A.; Landry, C.; Bellwon, P.; Kristl, T.; Hormann, K.; Ruzek, S.; Aiche, S.; et al. Evaluation of drug-induced neurotoxicity based on metabolomics, proteomics and electrical activity measurements in complementary CNS in vitro models. Toxicol. In Vitro 2015, 30, 138–165. [Google Scholar] [CrossRef] [PubMed]
- Illsinger, S.; Goken, C.; Brockmann, M.; Thiemann, I.; Bednarczyk, J.; Schmidt, K.H.; Lucke, T.; Hoy, L.; Janzen, N.; Das, A. Effect of tacrolimus on energy metabolism in human umbilical endothelial cells. Ann. Transplant. 2011, 16, 68–75. [Google Scholar] [CrossRef]
- Palacin, M.; Coto, E.; Llobet, L.; Pacheu-Grau, D.; Montoya, J.; Ruiz-Pesini, E. FK506 affects mitochondrial protein synthesis and oxygen consumption in human cells. Cell Biol. Toxicol. 2013, 29, 407–414. [Google Scholar] [CrossRef]
- Zini, R.; Simon, N.; Morin, C.; Thiault, L.; Tillement, J.P. Tacrolimus decreases in vitro oxidative phosphorylation of mitochondria from rat forebrain. Life Sci. 1998, 63, 357–368. [Google Scholar] [CrossRef]
- Jin, K.B.; Choi, H.J.; Kim, H.T.; Hwang, E.A.; Suh, S.I.; Han, S.Y.; Nam, S.I.; Park, S.B.; Kim, H.C.; Ha, E.Y.; et al. The production of reactive oxygen species in tacrolimus-treated glial cells. Transplant. Proc. 2008, 40, 2680–2681. [Google Scholar] [CrossRef]
- Jin, K.B.; Hwang, E.A.; Han, S.Y.; Park, S.B.; Kim, H.C.; Ha, E.Y.; Suh, S.I.; Mun, K.C. Effects of tacrolimus on antioxidant status and oxidative stress in glioma cells. Transplant. Proc. 2008, 40, 2740–2741. [Google Scholar] [CrossRef]
- Asai, A.; Qiu, J.; Narita, Y.; Chi, S.; Saito, N.; Shinoura, N.; Hamada, H.; Kuchino, Y.; Kirino, T. High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 1999, 274, 34450–34458. [Google Scholar] [CrossRef]
- Phillis, J.W.; Diaz, F.G.; O’Regan, M.H.; Pilitsis, J.G. Effects of immunosuppressants, calcineurin inhibition, and blockade of endoplasmic reticulum calcium channels on free fatty acid efflux from the ischemic/reperfused rat cerebral cortex. Brain Res. 2002, 957, 12–24. [Google Scholar] [CrossRef]
- Baumgartel, K.; Mansuy, I.M. Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 2012, 19, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J. FK506, an immunosuppressant targeting calcineurin function. Curr. Med. Chem. 2000, 7, 731–748. [Google Scholar] [CrossRef] [PubMed]
- De Weerdt, A.; Claeys, K.G.; De Jonghe, P.; Ysebaert, D.; Chapelle, T.; Roeyen, G.; Jorens, P.G. Tacrolimus-related polyneuropathy: Case report and review of the literature. Clin. Neurol. Neurosurg. 2008, 110, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Inui, K. An up-date review on individualized dosage adjustment of calcineurin inhibitors in organ transplant patients. Pharmacol. Ther. 2006, 112, 184–198. [Google Scholar] [CrossRef]
- Xu, X.; Su, B.; Barndt, R.J.; Chen, H.; Xin, H.; Yan, G.; Chen, L.; Cheng, D.; Heitman, J.; Zhuang, Y.; et al. FKBP12 is the only FK506 binding protein mediating T-cell inhibition by the immunosuppressant FK506. Transplantation 2002, 73, 1835–1838. [Google Scholar] [CrossRef]
- Lyons, W.E.; Steiner, J.P.; Snyder, S.H.; Dawson, T.M. Neuronal regeneration enhances the expression of the immunophilin FKBP-12. J. Neurosci. 1995, 15, 2985–2994. [Google Scholar] [CrossRef]
- Steiner, J.P.; Dawson, T.M.; Fotuhi, M.; Glatt, C.E.; Snowman, A.M.; Cohen, N.; Snyder, S.H. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature 1992, 358, 584–587. [Google Scholar] [CrossRef]
- Yokogawa, K.; Takahashi, M.; Tamai, I.; Konishi, H.; Nomura, M.; Moritani, S.; Miyamoto, K.; Tsuji, A. P-glycoprotein-dependent disposition kinetics of tacrolimus: Studies in mdr1a knockout mice. Pharm. Res. 1999, 16, 1213–1218. [Google Scholar] [CrossRef]
- Yu, J.J.; Zhang, Y.; Wang, Y.; Wen, Z.Y.; Liu, X.H.; Qin, J.; Yang, J.L. Inhibition of calcineurin in the prefrontal cortex induced depressive-like behavior through mTOR signaling pathway. Psychopharmacology (Berl) 2013, 225, 361–372. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, Y.H.; Wang, Y.X.; Liu, T.; Law, P.Y.; Loh, H.H.; Chen, H.Z.; Qiu, Y. Modulation of mTOR Activity by mu-Opioid Receptor is Dependent upon the Association of Receptor and FK506-Binding Protein 12. CNS Neurosci. Ther. 2015, 21, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T. Upstream and downstream of ran GTPase. Biol. Chem. 2000, 381, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Veroux, P.; Veroux, M.; Puliatti, C.; Morale, W.; Cappello, D.; Valvo, M.; Macarone, M. Tacrolimus-induced neurotoxicity in kidney transplant recipients. Transplant. Proc. 2002, 34, 3188–3190. [Google Scholar] [CrossRef]
- Henry, M.L. Cyclosporine and tacrolimus (FK506): A comparison of efficacy and safety profiles. Clin. Transplant. 1999, 13, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Ayas, M.; Al-Jefri, A.; Al-Seraihi, A. In cyclosporine induced neurotoxicity, is tacrolimus an appropriate substitute or is it out of the frying pan and into the fire? Pediatr. Blood Cancer 2008, 50, 426, author reply 427. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.Y.; Saadi, A.; Schmahmann, J.D. Pearls & Oy-sters: Tacrolimus neurotoxicity presenting as an isolated brainstem lesion. Neurology 2016, 87, 1423. [Google Scholar] [CrossRef]
- Arnold, R.; Pussell, B.A.; Pianta, T.J.; Lin, C.S.; Kiernan, M.C.; Krishnan, A.V. Association between calcineurin inhibitor treatment and peripheral nerve dysfunction in renal transplant recipients. Am. J. Transplant. 2013, 13, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Serkova, N.J.; Christians, U.; Benet, L.Z. Biochemical mechanisms of cyclosporine neurotoxicity. Mol. Interv. 2004, 4, 97–107. [Google Scholar] [CrossRef]
- Chen, C.C.; Hsu, L.W.; Huang, L.T.; Huang, T.L. Chronic administration of cyclosporine A changes expression of BDNF and TrkB in rat hippocampus and midbrain. Neurochem. Res. 2010, 35, 1098–1104. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Stress, Glucocorticoids, and Damage to the Nervous System: The Current State of Confusion. Stress 1996, 1, 1–19. [Google Scholar] [CrossRef]
- Venkataramanan, R.; Swaminathan, A.; Prasad, T.; Jain, A.; Zuckerman, S.; Warty, V.; McMichael, J.; Lever, J.; Burckart, G.; Starzl, T. Clinical pharmacokinetics of tacrolimus. Clin. Pharmacokinet. 1995, 29, 404–430. [Google Scholar] [CrossRef] [PubMed]
- Noe, A.; Cappelli, B.; Biffi, A.; Chiesa, R.; Frugnoli, I.; Biral, E.; Finizio, V.; Baldoli, C.; Vezzulli, P.; Minicucci, F.; et al. High incidence of severe cyclosporine neurotoxicity in children affected by haemoglobinopaties undergoing myeloablative haematopoietic stem cell transplantation: Early diagnosis and prompt intervention ameliorates neurological outcome. Ital. J. Pediatr. 2010, 36, 14. [Google Scholar] [CrossRef] [PubMed]
- Forgacs, B.; Merhav, H.J.; Lappin, J.; Mieles, L. Successful conversion to rapamycin for calcineurin inhibitor-related neurotoxicity following liver transplantation. Transplant. Proc. 2005, 37, 1912–1914. [Google Scholar] [CrossRef]
- Chohan, R.; Vij, R.; Adkins, D.; Blum, W.; Brown, R.; Tomasson, M.; Devine, S.; Graubert, T.; Goodnough, L.T.; DiPersio, J.F.; et al. Long-term outcomes of allogeneic stem cell transplant recipients after calcineurin inhibitor-induced neurotoxicity. Br. J. Haematol. 2003, 123, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, I.; Schmauss, D.; Sodian, R.; Beiras-Fernandez, A.; Oberhoffer, M.; Daebritz, S.; Schoenberg, S.O.; Reichart, B. Late-onset tacrolimus-associated cerebellar atrophia in a heart transplant recipient. J. Heart Lung Transplant. 2007, 26, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Froud, T.; Baidal, D.A.; Ponte, G.; Ferreira, J.V.; Ricordi, C.; Alejandro, R. Resolution of neurotoxicity and beta-cell toxicity in an islet transplant recipient following substitution of tacrolimus with MMF. Cell Transplant. 2006, 15, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Daoud, D.; Scheld, H.H.; Speckmann, E.J.; Gorji, A. Rapamycin: Brain excitability studied in vitro. Epilepsia 2007, 48, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.A.; Hategan, A. Immunosuppressant-associated neurotoxicity responding to olanzapine. Case Rep. Psychiatry 2014, 2014, 250472. [Google Scholar] [CrossRef]
- O’Donnell, M.M.; Williams, J.P.; Weinrieb, R.; Denysenko, L. Catatonic mutism after liver transplant rapidly reversed with lorazepam. Gen. Hosp. Psychiatry 2007, 29, 280–281. [Google Scholar] [CrossRef]
- Ungvari, G.S.; Chiu, H.F.; Chow, L.Y.; Lau, B.S.; Tang, W.K. Lorazepam for chronic catatonia: A randomized, double-blind, placebo-controlled cross-over study. Psychopharmacology (Berl) 1999, 142, 393–398. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Makuuchi, M.; Harihara, Y.; Imamura, H.; Sato, H. Correlation between neurotoxic events and intracerebral concentration of tacrolimus in rats. Biol. Pharm. Bull. 2000, 23, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Makuuchi, M.; Harihara, Y.; Imamura, H.; Sato, H. Higher intracerebral concentration of tacrolimus after intermittent than continuous administration to rats. Liver Transpl. 2001, 7, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Oishi, R.; Kataoka, Y. Tacrolimus-induced neurotoxicity and nephrotoxicity is ameliorated by administration in the dark phase in rats. Cell Mol. Neurobiol. 2004, 24, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Spallanzani, V.; Bindi, L.; Bianco, I.; Precisi, A.; DeSimone, P.; Mazzoni, A.; Biancofiore, G. Red blood cell exchange as an approach for treating a case of severe tacrolimus overexposure. Transfus Apher. Sci. 2017, 56, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Enokida, H.; Nagano, S.; Yokouchi, M.; Hayami, H.; Komiya, S.; Nakagawa, M. Effects of blood purification therapy on a patient with ifosfamide-induced neurotoxicity and acute kidney injury. J. Artif. Organs 2014, 17, 110–113. [Google Scholar] [CrossRef]
- Drake, M.; Friberg, H.; Boris-Moller, F.; Sakata, K.; Wieloch, T. The immunosuppressant FK506 ameliorates ischaemic damage in the rat brain. Acta Physiol. Scand. 1996, 158, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, J.; Butcher, S.P. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature 1994, 371, 336–339. [Google Scholar] [CrossRef]
- Macleod, M.R.; Butcher, S.P. Nitric-oxide-synthase-mediated cyclic guanosine monophosphate production in neonatal rat cerebellar prisms is resistant to calcineurin inhibition. Neurosci. Lett. 2002, 322, 41–44. [Google Scholar] [CrossRef]
- Dethloff, T.; Hansen, B.A.; Larsen, F.S. Tacrolimus ameliorates cerebral vasodilatation and intracranial hypertension in the rat with portacaval anastomosis and hyperammonemia. Liver Transpl. 2004, 10, 922–927. [Google Scholar] [CrossRef]
- Arora, R.B.; Kumar, K.; Deshmukh, R.R. FK506 attenuates intracerebroventricular streptozotocin-induced neurotoxicity in rats. Behav. Pharmacol. 2013, 24, 580–589. [Google Scholar] [CrossRef]
- Nishimura, T.; Imai, H.; Minabe, Y.; Sawa, A.; Kato, N. Beneficial effects of FK506 for experimental temporal lobe epilepsy. Neurosci. Res. 2006, 56, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Kay, K.; Matsouka, R.; Rozkalne, A.; Betensky, R.A.; Hyman, B.T. Calcineurin inhibition with systemic FK506 treatment increases dendritic branching and dendritic spine density in healthy adult mouse brain. Neurosci. Lett. 2011, 487, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Yanagidaira, T.; Takahashi, Y.; Sano, K.; Murata, T.; Hayashi, T. Development of spontaneous neuropathy in NF-kappaBp50-deficient mice by calcineurin-signal involving impaired NF-kappaB activation. Mol. Vis. 2011, 17, 2157–2170. [Google Scholar] [PubMed]
- Zawadzka, M.; Kaminska, B. Immunosuppressant FK506 affects multiple signaling pathways and modulates gene expression in astrocytes. Mol. Cell Neurosci. 2003, 22, 202–209. [Google Scholar] [CrossRef]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Dose-dependent, protective effect of FK506 against white matter changes in the rat brain after chronic cerebral ischemia. Brain Res. 1998, 792, 105–113. [Google Scholar] [CrossRef]
- Bultynck, G.; De Smet, P.; Weidema, A.F.; Ver Heyen, M.; Maes, K.; Callewaert, G.; Missiaen, L.; Parys, J.B.; De Smedt, H. Effects of the immunosuppressant FK506 on intracellular Ca2+ release and Ca2+ accumulation mechanisms. J. Physiol. 2000, 525(Pt. 3), 681–693. [Google Scholar] [CrossRef]
- Hansson, M.J.; Persson, T.; Friberg, H.; Keep, M.F.; Rees, A.; Wieloch, T.; Elmer, E. Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 2003, 960, 99–111. [Google Scholar] [CrossRef]
- Yardin, C.; Terro, F.; Lesort, M.; Esclaire, F.; Hugon, J. FK506 antagonizes apoptosis and c-jun protein expression in neuronal cultures. Neuroreport. 1998, 9, 2077–2080. [Google Scholar] [CrossRef]
- Koike, K.; Hashimoto, K.; Fukami, G.; Okamura, N.; Zhang, L.; Ohgake, S.; Koizumi, H.; Matsuzawa, D.; Kawamura, N.; Shimizu, E.; et al. The immunophilin ligand FK506 protects against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum. Neuropharmacology 2005, 48, 391–397. [Google Scholar] [CrossRef]
- Kaminska, B.; Gaweda-Walerych, K.; Zawadzka, M. Molecular mechanisms of neuroprotective action of immunosuppressants--facts and hypotheses. J. Cell Mol. Med. 2004, 8, 45–58. [Google Scholar] [CrossRef]
- Santos, J.B.; Schauwecker, P.E. Protection provided by cyclosporin A against excitotoxic neuronal death is genotype dependent. Epilepsia 2003, 44, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.W.; Kroner, A.; Kohl, B.; Wessig, C.; Martini, R. Tacrolimus (FK506) causes disease aggravation in models for inherited peripheral myelinopathies. Neurobiol. Dis. 2009, 33, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Setkowicz, Z.; Guzik, R. Injections of vehicle, but not cyclosporin A or tacrolimus (FK506), afford neuroprotection following injury in the developing rat brain. Acta Neurobiol. Exp. (Wars) 2007, 67, 399–409. [Google Scholar] [PubMed]
- Pignataro, G.; Capone, D.; Polichetti, G.; Vinciguerra, A.; Gentile, A.; Di Renzo, G.; Annunziato, L. Neuroprotective, immunosuppressant and antineoplastic properties of mTOR inhibitors: Current and emerging therapeutic options. Curr. Opin. Pharmacol. 2011, 11, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, A. Therapeutic approaches to epileptogenesis--hope on the horizon. Epilepsia 2010, 51 (Suppl. 3), 2–17. [Google Scholar] [CrossRef]
- Carloni, S.; Mazzoni, E.; Cimino, M.; De Simoni, M.G.; Perego, C.; Scopa, C.; Balduini, W. Simvastatin reduces caspase-3 activation and inflammatory markers induced by hypoxia-ischemia in the newborn rat. Neurobiol. Dis. 2006, 21, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Kim, H.H.; Hiroi, Y.; Sawada, N.; Salomone, S.; Benjamin, L.E.; Walsh, K.; Moskowitz, M.A.; Liao, J.K. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci. Signal. 2009, 2, ra11. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.E.; Shin, I.C.; Koh, H.C. Autophagy regulates chlorpyrifos-induced apoptosis in SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2013, 268, 55–67. [Google Scholar] [CrossRef]
- Chen, L.; Hu, L.; Dong, J.Y.; Ye, Q.; Hua, N.; Wong, M.; Zeng, L.H. Rapamycin has paradoxical effects on S6 phosphorylation in rats with and without seizures. Epilepsia 2012, 53, 2026–2033. [Google Scholar] [CrossRef]
- Saliba, S.W.; Vieira, E.L.; Santos, R.P.; Candelario-Jalil, E.; Fiebich, B.L.; Vieira, L.B.; Teixeira, A.L.; de Oliveira, A.C. Neuroprotective effects of intrastriatal injection of rapamycin in a mouse model of excitotoxicity induced by quinolinic acid. J. Neuroinflamm. 2017, 14, 25. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Corticosteroids | |
|---|---|
| Generic Name | Prednisone; Prednisolone; Methylprednisolone; Dexamethasone |
| Trade Name | Prelone®, Orapred®, Millipred®, Orapred ODT®; Prednisol®, Pred Forte®, Pred Mild®, Omnipred®; Medrol®, Medrol Dosepak®, MethylPREDNISolone Dose Pack®, Solu-Medrol®; Decadron®, Dexamethasone Intensol®, Dexasone®, Hexadrol® |
| Mechanism of Action | The mechanisms of action are diverse and include interference with intracellular transcription factors and signaling pathways of several surface receptors, including the T cell antigen receptor and downstream kinases, thereby blocking the transcription of cytokine genes and inhibiting cytokine production by T cells and macrophages [32]. |
| Role in Therapy | Maintenance; high doses of corticosteroids (>1 mg/kg), used for induction therapy in transplantation; treatment of acute cellular rejection and AMR [3,32]. |
| Adverse Effects | Hypertension, hyperlipidemia, glucose intolerance, malignancy, Cushingoid features, sleep disturbances, mood changes, impaired wound healing, osteoporosis, psychosis, photosensitivity, acne hirsutism, avascular necrosis, weight gain, fluid retention, increased appetite, menstrual irregularities, growth inhibition, GI disturbance, cataracts, infection [3]. |
| Monitoring Parameters | Glucose, blood pressure, fasting lipid panel, weight, DEXA scan, eye exam, intensive organ function monitoring [31]. |
| Other Information | Their role in the maintenance of immunosuppression is under investigation because of severe side effects during long-term use, but an immunosuppressive strategy without steroids could be only tried in low immunological risk transplant recipients; it also seems that treatment of steroids 1 h prior to ATG preoperatively may minimize CRS [3,4]. |
| Purine synthesis inhibitors | |
| Generic Name | Azathioprine; Mycophenolate mofetil; Mycophenolate sodium; Cyclophosphamide |
| Trade Name | Imuran®; Cellcept®; Myfortic®; Cytoxan®, Neosar®, Endoxan® |
| Mechanism of Action | Two distinct mechanisms participate in the inhibition of de novo DNA synthesis block cell division and then block cell division. AZA is a prodrug for 6-mercaptopurine, Mycophenolate mofetil is a prodrug of MPA and Mycophenolate sodium is an enteric-coated formulation of MPA. AZA blocks purine synthesis enzymes by incorporating into newly synthetized DNA and, finally, impedes DNA and RNA synthesis [31]. MPA selectively and noncompetitively inhibits a key enzyme in the de novo synthesis of purine named IMPDH and thus, inhibits proliferation of T and B lymphocyte [32]. |
| Role in Therapy | Maintenance |
| Adverse Effects | AZA: Hepatotoxicity, bone marrow suppression, malignancies (high dosages), macrocytic anemia, GI disturbance, alopecia, pancreatitis, infections [31]; MMF and Mycophenolate sodium: Dyslipidemia, DM, infections, bone marrow suppression, GI symptoms and anemia are common, while nephrotoxicity, neurotoxicity, and hepatotoxicity are uncommon [18,30]; CP: Low blood count, alopecia, GI symptoms, poor appetite, discoloration of the skin or nails. |
| Monitoring Parameters | AZA: CBC, LFT, amylase, lipase, TPMT enzyme level; MMF and Mycophenolate sodium: CBC, REMS; CP: CBC, LFT, KFT [31]. |
| Other Information | Newer trials have shown that AZA and MMF have similar efficacy. Low or absent TPMT activity is associated with increased AZA-associated myelosuppression. MPA is associated with pregnancy loss and congenital malformations when used during pregnancy. MPA may be of special interest in preventing the rise of DSA titers in pre-sensitized recipients. Patients with renal dysfunction need dosage adjustment when using MPA [28,31]. CP is associated with pregnancy loss and congenital malformations when used during pregnancy. |
| CNIs | |
| Generic Name | Tacrolimus; Cyclosporine |
| Trade Name | Prograf®, Graceptor®, Advagraf®, Envarsus XR®, Astagraf XL®; Neoral®, Gengraf®, Sandimmune® |
| Mechanism of Action | CNIs block signal transduction by activated NFAT through two distinct mechanisms. Tacrolimus binds to FKBP12 while CsA in combination of cyclophilin inhibits calcineurin-mediated dephosphorylation of NFAT, ultimately preventing cytokine transduction including IL-2 and IFNγ and T cell activation. In humoral immune response, CNIs interfere with T helper signals rather than targeting B cell directly [32]. |
| Role in Therapy | Maintenance |
| Adverse Effects | Often dose- and concentration- dependent, nephrotoxicity, infections, hyperkalemia, hypomagnesemia, hyperuricemia, cholelithiasis, GI symptoms, malignancy; tacrolimus > CsA: insulin-dependent diabetes mellitus, neurotoxicity; CsA > tacrolimus: hypertension, hypercholesterolemia, hyperlipidemia; CsA only: gingival hyperplasia, hirsutism; tacrolimus only: alopecia [3,4,31]. |
| Monitoring Parameters | Trough levels, serum creatinine, potassium, magnesium, uric acid [31] |
| Other Information | Tacrolimus seems more effective than CsA-based immunosuppressive regimens, so tacrolimus-based immunosuppression usually used as a first-line therapy after transplantation. Tacrolimus is metabolized by CYP3A and has potential drug interactions. Neurotoxicity more likely occurs in liver transplant patients with low serum cholesterol levels. Patients with hepatic dysfunction or advanced age have high risk of drug interactions after CSA [3,4,18,31]. |
| mTOR inhibitors | |
| Generic Name | Sirolimus (Rapamycin); Everolimus; |
| Trade Name | Rapamune®; Certican®, Zortress® |
| Mechanism of Action | These drugs in combination of FKBP12 inhibit mTOR and impede the translation of mRNA-encoding proteins which are necessary to the cell cycle, thus reducing IL-2-mediated T cell proliferation and cytokine production. In contrast to CNIs, they seem to do not influence the early phase of T-cell activation [31,32]. |
| Role in Therapy | Maintenance |
| Adverse Effects | Dyslipidemia, mucositis, edema, proteinuria, wound-related reactions, mouth ulcers, bone pain, diarrhea, pneumonitis, venous thromboembolism, infections, low blood count [3] |
| Monitoring Parameters | Trough levels, fasting lipid panel, CBC, LFT [31] |
| Other Information | Only sirolimus is reported to have direct inhibitory effects on the proliferation of B cells and their differentiation into plasma cells [32]. An mTOR inhibitor–based regimen is under investigation for low risk of nephrotoxicity or neurotoxicity when used alone [3]. |
| Monoclonal antibodies | |
| Generic Name | Muromonab-CD3; Rituximab; Basiliximab; Daclizumab; Alemtuzumab; Eculizumab |
| Trade Name | Orthoclone OKT3®; Rituxan®; Simulect®; Zinbryta®; Campath®, Lemtrada®; Soliris® |
| Mechanism of Action | Muromonab-CD3: first monoclonal antibody approved for use in solid-organ transplantation, direct against the CD3 marker on all mature human T cells [30]. Rituximab: a murine/human chimeric monoclonal antibody directly targets the CD20 surface marker on B cells [33]. Basiliximab: a murine/human chimeric monoclonal antibody competitively inhibits CD25 complex, the alpha subunit of the IL-2 receptor which present only on activated and non-resting T cell, thereby inhibiting T cell proliferation [34]. Daclizumab: a humanized monoclonal antibody similar to Basiliximab, has high specificity and affinity against CD25 complex [34]. Alemtuzumab: a recombinant DNA-derived, humanized anti-CD52 monoclonal antibody targets T and B lymphocytes, NK cells, monocytes, and macrophages, finally leading to rapid and powerful depletion of T and B lymphocytes, and monocytes [31]. Eculizumab: a humanized monoclonal antibody binds to complement C5 with high affinity and blocks complement cascade by preventing the formation of the terminal membrane attack complex [34]. |
| Role in Therapy | Muromonab-CD3: withdrawn Rituximab: Desensitization, treatment of AMR, and for cases of PTLD [30,31] Basiliximab, Daclizumab: Induction Alemtuzumab: Induction, treatment of AMR and steroid-resistant rejection [31] Eculizumab: Desensitization, treatment of AMR [3] |
| Adverse Effects | Muromonab-CD3: Serious CRS Rituximab: Bone marrow suppression, infusion-related events [3] Basiliximab: Rare; infections, bone marrow suppression, hypersensitivity reactions [3] Daclizumab: GI disturbance, rare lymphoproliferative disorders and malignancies [33] Alemtuzumab: Bone marrow suppression, infusion reaction, infections, mild CRS, headaches, induction of autoimmune disease, a possible increased risk of PTLD [31,34] Eculizumab: Increased risk for gram-negative bacterial infection, bone marrow suppression [3,30] |
| Monitoring Parameters | Alemtuzumab: Vital signs, CBC, absolute lymphocyte count [31] |
| Other Information | Rituximab: Has been tested as an induction agent in cell therapy [3]. Basiliximab: Induction therapy using basiliximab has higher rejection rates [3]. Alemtuzumab: Usage in induction and acute rejection treatment is still under study; has a similar immunosuppressive effect to ATG, but less side effects. Pre-treatment of diphenhydramine and acetaminophen can decrease side effects [30,31]. Eculizumab: The usage for immunosuppressants has been only reported in case reports and observational studies, has limited efficacy and high cost [34]. |
| Polyclonal antibodies | |
| Generic Name | Antithymocyte globulin |
| Trade Name | Thymoglobulin® |
| Mechanism of Action | This drug depletes the number of circulating T lymphocytes by antibody–dependent cell–mediated or complement-depend cytotoxicity and their interaction with T cell surface antigens, may result in apoptosis, which alters T cell activation and homing [32]. |
| Role in Therapy | Induction; treatment of steroid-resistant rejection [3] |
| Adverse Effects | Malignancies, infections, bone marrow suppression, CRS, pulmonary edema, phlebitis, pruritis, erythema, serum sickness [3,31] |
| Monitoring Parameters | White blood cells, platelet count, vital signs, CD3 count [31] |
| Other Information | To prevent an intense CRS, pre-treatment with systemic glucocorticoids, antihistamines and antipyretics should precede drug administration; preferred in sensitized patients without DSAs [31,32]. |
| Co-stimulation blockade agent | |
| Generic Name | Belatacept |
| Trade Name | Nulojix® |
| Mechanism of Action | An agent mimics soluble CTLA-4 and binds to CD86/80 on APCs, thus blocking T-cell activation. Moreover, it maybe indirectly prevent production of antigen-specific antibody (IgG, IgM, and IgA) by B lymphocytes or directly affect B lymphocytes [30,32]. |
| Role in Therapy | Induction; maintenance |
| Adverse Effects | Malignancies, bone marrow suppression, diarrhea, infection, edema, hypertension, dyslipidemia, DM, proteinuria, electrolyte disorders, dyspnea, purpura, transaminitis, temporal lobe epilepsy. More than 20% of patients experience side effects [31,34]. |
| Monitoring Parameters | EBV serostatus (prior to treatment) [31] |
| Other Information | Only used for adult patients; no drug-drug interactions; patients with renal or hepatic impairment need no dosage adjustment; contraindicated in recipients who are EBV seronegative or with unknown EBV serostatus [4,30,31]. |
| Immunosuppressants in development | |
| Generic Name | FK778; Tofacitinib (CP-690550); Bortezomib (PS341); Tocilizumab; IdeS (imlifidase); Fingolimod (FTY720); Alefacept; ASKP1240; Voclosporin (ISA247); Sotrastaurin (AEB071); Siplizumab; TOL101; Efalizumab; Belimumab; Sutimlimab (BIVV009); C1-INH (C1 esterase inhibitor) |
| Trade Name | none; Xeljanz®; Velcade®; Actemra®; none; Gilenya®; Amevive®; none; Luveniq®; none; none; none; Raptiva®, Genentech®, Merck Serono®; Benlysta®; none; Berinert®, Cinryze®, Haegarda® |
| Mechanism of Action | FK778: An agent blocks pyrimidine synthesis by blockade of DHODH and inhibition of tyrosine kinase activity, thus inhibiting both T-cell and B-cell function; moreover, it can directly inhibit lymphocyte activation, attenuate lymphocyte-endothelium interactions [4,28]. Tofacitinib: A JAK3 inhibitor, that exerts its effects by uncoupling cytokine receptor signaling from downstream STAT transcriptional activation and subsequently, suppressing various cytokine-regulated signaling, thus influencing lymphocyte activation, proliferation, differentiation, and function [4,29,33]. Bortezomib: A reversible 26S proteasome inhibitor that can delete non-transformed plasma cells, which is critical to alloantibodies [30]. Tocilizumab: A first-in-class, humanized, monoclonal antibody directed against IL-6R [29,33]. IdeS: An enzyme from Streptococcus pyogenes that specifically cleaves human IgG antibodies [33]. Fingolimod: A structural analogue of sphingosine, metabolized by sphingosine kinases to fingolimod-phosphate in the cell; this active metabolite can entrap lymphocytes in secondary lymphoid organs and reduce their number in peripheral blood, thus reducing cell-mediated immune responses [4,28]. Alefacept: Directed against the extracellular CD2 receptor expressed on T lymphocytes thus inhibiting lymphocyte activation and production; blocks the CD2/LFA-3 interaction and impedes helper T-cell adhesion to APCs [29,34]. ASKP1240: A novel, fully human anti-CD40 monoclonal antibody, is currently under study in phase II clinical trials in kidney transplantation [29]. Voclosporin: A novel oral semisynthetic analogue of CsA, inhibits calcineurin [29]. Sotrastaurin: An oral protein kinase C inhibitor that can block T-cell activation [29]. Siplizumab: A novel humanized monoclonal antibody, binds to CD2 antigen on T lymphocyte or NK cell [29]. TOL101: A highly selective murine monoclonal antibody targeting the αβ-TCR [29]. Efalizumab: An anti-lymphocyte function-associated antigen molecule that inhibits lymphocyte activation and migration [29]. Belimumab: A human monoclonal antibody that inhibits BAFF [29]. Sutimlimab: Selectively blocks the classical pathway of complement -specific serine protease C1s to prevent the formation of the classic C3 convertase pathway [29]. C1-INH: A serine-protease inhibitor inhibits complement system by binding to and inactivating C1r and C1s and dissociating the C1 complex [29]. |
| Role in THERAPY | FK778: Further development for the treatment of transplantation has been discontinued [4,28]. Tofacitinib: Withdrawn in transplantation. Bortezomib: Desensitization, treatment of AMR [30]. Tocilizumab: Desensitization [29] IdeS: Desensitization [33] Fingolimod: No further development for the treatment of transplantation [4,28]. Alefacept: Withdrawn in transplantation. ASKP1240: Immunosuppressive effects in nonhuman primates have been proven [29]. Voclosporin: Its efficacy in preventing acute rejection is as potent as tacrolimus by a phase 2b PROMISE study [29]. Sotrastaurin: May be an alternative therapy for Cis [29]. Siplizumab: Has been tested as an induction drug in a human study [29]. TOL101: Has been tested as an induction agent to prevent rejection is currently under study in phase II clinical trials [29]. Efalizumab: Withdrawn. Belimumab: The usage as supplement to standard-of-care immunosuppressive therapy in renal transplantation has been proven by a phase II study [29]. Sutimlimab: A single-arm pilot trial showed that BIVV009 effectively blocks the alloantibody-triggered classical pathway activation in kidney transplant recipients [29]. C1-INH: The results of a recent placebo-controlled trial suggested that C1-INH replacement may be useful in the treatment of AMR [29]. |
| Adverse Effects | Tofacitinib: Infection, CMV disease, PTLD, anemia, neutropenia [29]. Bortezomib: GI syndromes, asthenia, neurotoxicity, bone marrow suppression, shingles [3,30]. Tocilizumab: Infections. Fingolimod: Bradycardia, macular oedema, increased airway resistance, a “first-dose” negative chronotropic effect [4,28]. Alefacept: Malignancies. Sotrastaurin: GI events are common [29]. Efalizumab: Infections, PML, PTLD. Belimumab: Infection, hypersensitivity, malignancy. |
| Monitoring Parameters | Tofacitinib: Drug serum levels [4,29,33]. |
| Other Information | FK778: There have been no results proving the efficacy of FK778 in phase III studies. Therefore, its development was been discontinued for organ transplantation in 2006 [29]. Tofacitinib: When combined with MMF, the rates of viral infection and viral-associated malignancies may increase [29]. Bortezomib: Small, non-randomised trials suggest efficacy in AMR; may decrease AMR in highly sensitised individuals [30]. IdeS: IdeS has been proven to effectively reduce anti-HLA antibody levels in highly sensitized patients by a phase II study; clinical trials in sensitized kidney patients are ongoing [29]. Fingolimod: It is now approved for use in MS, but its mechanism is still unknown. Alefacept: Its use for the prevention of graft-versus host disease is under investigation. ASKP1240: Further clinical III studies are needed [29]. Voclosporin: Low-dose voclosporin may reduce incidence of new-onset diabetes after transplantation [29]. Sotrastaurin: High-dose sotrastaurin may be associated with faster heart rates [29]. Sutimlimab: Undergoing phase clinical III trail [29]. C1-INH: Further studies are needed to confirm the safety and efficacy of C1-INH in the treatment of AMR [29]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Egashira, N.; Masuda, S. Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. Int. J. Mol. Sci. 2019, 20, 3210. https://doi.org/10.3390/ijms20133210
Zhang W, Egashira N, Masuda S. Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. International Journal of Molecular Sciences. 2019; 20(13):3210. https://doi.org/10.3390/ijms20133210
Chicago/Turabian StyleZhang, Wei, Nobuaki Egashira, and Satohiro Masuda. 2019. "Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities" International Journal of Molecular Sciences 20, no. 13: 3210. https://doi.org/10.3390/ijms20133210
APA StyleZhang, W., Egashira, N., & Masuda, S. (2019). Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. International Journal of Molecular Sciences, 20(13), 3210. https://doi.org/10.3390/ijms20133210