Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases

 , , , , and
, , , , and 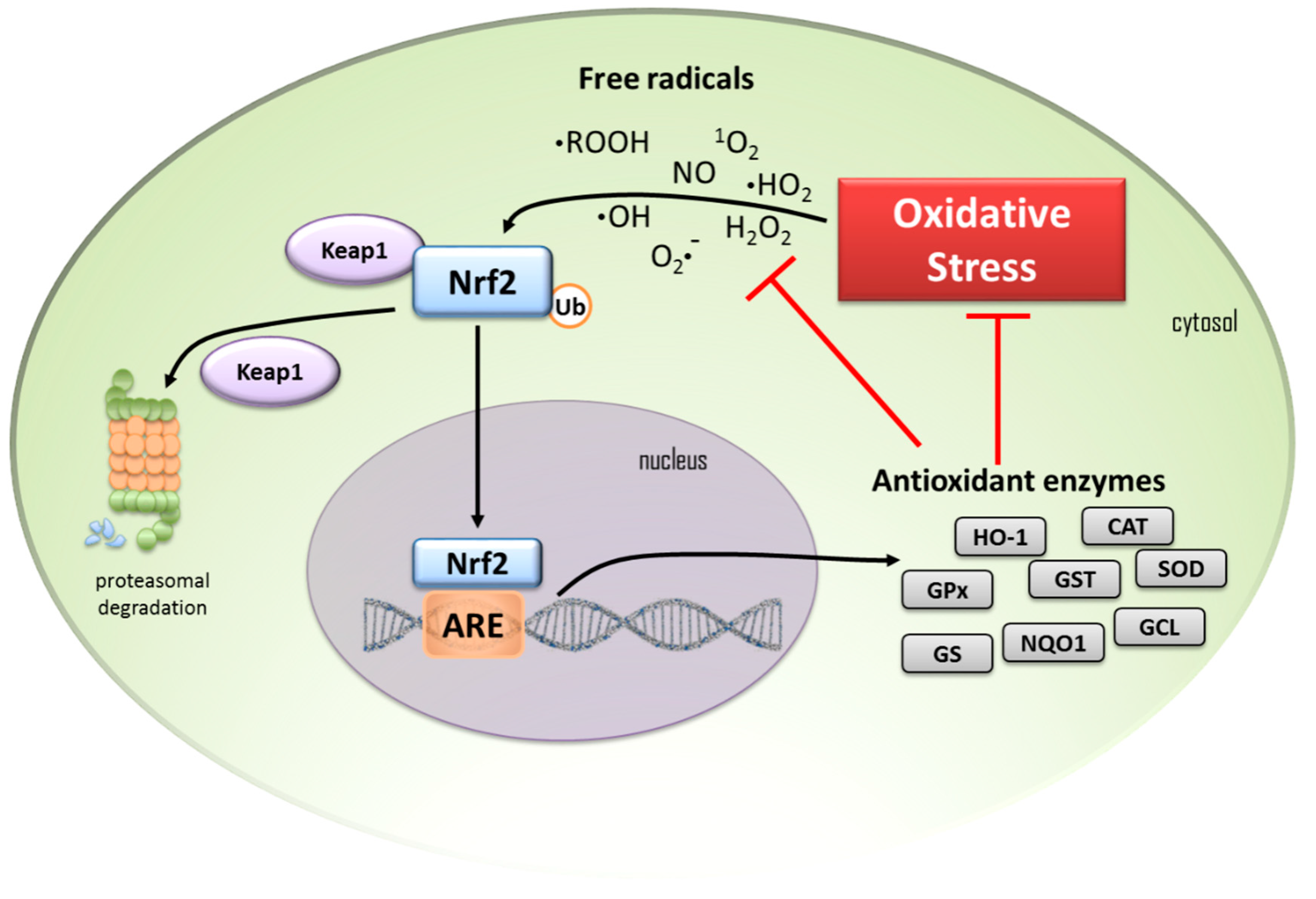{kind=link}
{kind=link}
Abstract
1. Introduction
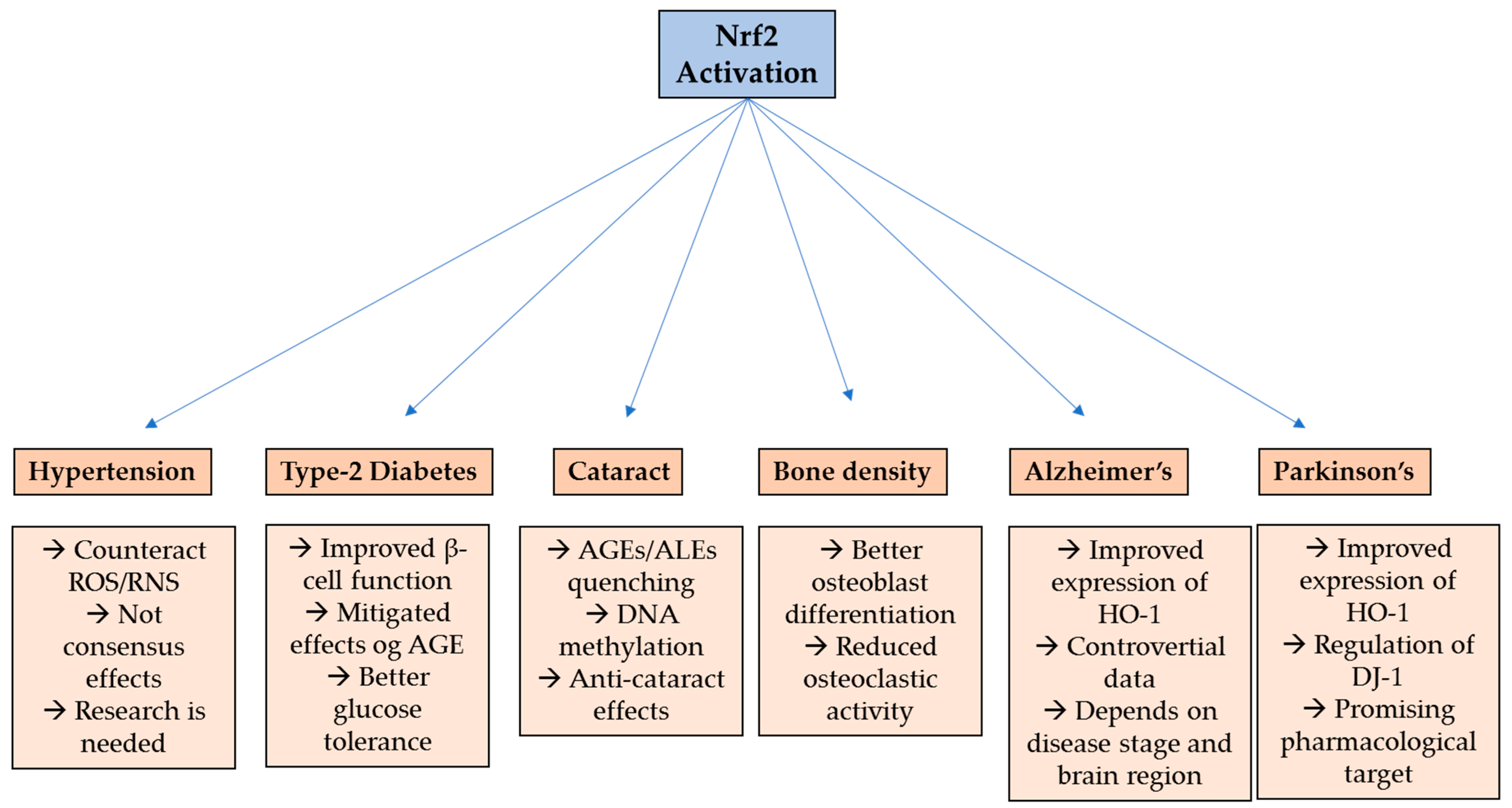
2. Hypertension
3. Type-2 Diabetes
4. Cataract
5. Bone Metabolism Disorders
6. Alzheimer’s Disease
7. Parkinson’s Disease
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cai, H.; Cong, W.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic Dysfunction in Alzheimer’s Disease and Related Neurodegenerative Disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Francisqueti, F.V.; Chiaverini, L.C.T.; dos Santos, K.C.; Minatel, I.O.; Ronchi, C.B.; Ferron, A.J.T.; Ferreira, A.L.A.; Correa, C.R. The role of oxidative stress on the pathophysiology of metabolic syndrome. Rev. Assoc. Med. Bras. 2017, 63, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2016, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Jakóbkiewicz-Banecka, J.; Kloska, A.; Smolińska, E.; Mozolewski, P.; Malinowska, M.; Rychlowski, M.; Banecki, B.; Wegrzyn, G.; Gabig-Cimińska, M. Modulation of expression of genes involved in glycosaminoglycan metabolism and lysosome biogenesis by flavonoids. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Yamamoto, M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002, 348, 182–190. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Pickett, B.C. Transcriptional Regulation of the Rat Glutathione S-Transferase Ya Subunit Gene. J. Biol. Chem. 1990, 265, 14648–14653. [Google Scholar]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Biochemistry 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bolívar, J.J. Essential Hypertension: An Approach to Its Etiology and Neurogenic Pathophysiology. Int. J. Hypertens. 2013, 2013, 547809. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E. Contribution of mitochondrial oxidative stress to hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; De Luca, N.; Trimarco, B.; Iaccarino, G. The antioxidant therapy: New insights in the treatment of hypertension. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ramon, R.; Gonza, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef]
- Brito, R.; Castillo, G.; Valls, N.; Rodrigo, R.; Program, C.P. Oxidative Stress in Hypertension: Mechanisms and Therapeutic Opportunities. Exp. Clin. Endocrinol. Diabetes 2015, 123, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.A.; Runge, M.S.; Madamanchi, N.R. Oxidative stress, NADPH oxidases, and arteries. Hamostaseologie 2016, 36, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Barancik, M.; Gresova, L.; Barteková, M.; Dovinová, I. Nrf2 as a Key Player of Redox Regulation in Cardiovascular Diseases. Physiol. Res. 2016, 65, S1–S10. [Google Scholar] [PubMed]
- Lopes, R.A.; Neves, K.B.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 2015, 66, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Care, D.; Suppl, S.S. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42, 13–28. [Google Scholar] [CrossRef]
- World Health Organization. Diabetes Mellitus 2013, No. 138. Available online: https://www.who.int/mediacentre/factsheets/fs138/en/ (accessed on 5 March 2019).
- Cruz-Jentoft, A.J.; Carpena-Ruiz, M.; Montero-Errasquín, B.; Sánchez-Castellano, C.; Sánchez-García, E. Exclusion of older adults from ongoing clinical trials about type 2 diabetes mellitus. J. Am. Geriatr. Soc. 2013, 61, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Cowie, C.C.; Rust, K.F.; Ford, E.S.; Eberhardt, M.S.; Byrd-Holt, D.D.; Li, C.; Williams, D.E.; Gregg, E.W.; Bainbridge, K.E.; Saydah, S.H.; et al. Full accounting of diabetes and pre-diabetes in the, U.S. population in 1988–1994 and 2005–2006. Diabetes Care 2009, 32, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications IR RAGE SOD. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islet compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Ramkumar, K.; Vijayakumar, R.; Vanitha, P.; Suganya, N.; Manjula, C.; Rajaguru, P.; Sivasubramanian, S.; Gunasekaran, P. Protective effect of gallic acid on alloxan-induced oxidative stress and osmotic fragility in rats. Hum. Exp. Toxicol. 2014, 33, 638–649. [Google Scholar] [CrossRef]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 2010, 244, 77–83. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2017, 29, 1727–1745. [Google Scholar] [CrossRef]
- Bhakkiyalakshmi, E.; Sireesh, D.; Rajaguru, P.; Paulmurugan, R.; Ramkumar, K.M. The emerging role of redox-sensitive Nrf2-Keap1 pathway in diabetes. Pharmacol. Res. 2015, 91, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 System Prevents Onset of Diabetes Mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.K.; Reddy, V.T.; Konopleva, M.; Andreeff, M.; Chan, L. The Triterpenoid 2-Cyano-3,12-dioxooleana-1,9-dien-28-oic-acid Methyl Ester Has Potent Anti-diabetic Effects in Diet-induced Diabetic Mice and Lepr db/db Mice. J. Biol. Chem. 2010, 285, 40581–40592. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, Y.M.; Kim, J.W.; Kim, S.G. Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology 2007, 46, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.I.A.; Souza, E.M.D.; Pedrosa, F.D.O.; Réa, R.R.; Alves, A.D.S.C.; Picheth, G.; Rego, F.G.D.M. RAGE receptor and its soluble isoforms in diabetes mellitus complications. J. Bras. Patol. Med. Lab. 2013, 49, 97–108. [Google Scholar] [CrossRef]
- Marcovecchio, M.L.; Lucantoni, M.; Chiarelli, F. Role of Chronic and Acute Hyperglycemia in the Development of Diabetes Complications. Diabetes Technol. Ther. 2011, 13, 389–394. [Google Scholar] [CrossRef]
- Hsu, W.H.; Lee, B.H.; Chang, Y.Y.; Hsu, Y.W.; Pan, T.M. A novel natural Nrf2 activator with PPARγ-agonist (monascin) attenuates the toxicity of methylglyoxal and hyperglycemia. Toxicol. Appl. Pharmacol. 2013, 272, 842–851. [Google Scholar] [CrossRef]
- Geoffrion, M.; Du, X.; Irshad, Z.; Vanderhyden, B.C.; Courville, K.; Sui, G.; D’Agati, V.D.; Ott-Braschi, S.; Rabbani, N.; Thornalley, P.J.; et al. Differential effects of glyoxalase 1 overexpression on diabetic atherosclerosis and renal dysfunction in streptozotocin-treated, apolipoprotein E-deficient mice. Physiol. Rep. 2014, 2, e12043. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; McDonald, P.R.; Liu, J.; Klaassen, C.D. Screening of Natural Compounds as Activators of the Keap1-Nrf2 Pathway. Planta Med. 2014, 80, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Osorio, A.S.; González-Reyes, S.; Pedraza-Chaverri, J. Natural Nrf2 activators in diabetes. Clin. Chim. Acta 2015, 25, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Uruno, A.; Dv, C.; Tw, K.; Yamamoto, M. Nrf2 represses the onset of type 1 diabetes in non-obese diabetic mice. J. Endocrinol. 2019. [Google Scholar] [CrossRef]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef]
- Tastekin, B.; Pelit, A.; Polat, S.; Tuli, A.; Sencar, L.; Alparslan, M.M.; Daglioglu, Y.K. Therapeutic Potential of Pterostilbene and Resveratrol on Biomechanic, Biochemical, and Histological Parameters in Streptozotocin-Induced Diabetic Rats. Evid.-Based Complement Altern. Med. 2018, 9012352, 1–10. [Google Scholar] [CrossRef]
- Nourooz-Zadeh, J.; Pereira, P. Age-related accumulation of free polyunsaturated fatty acids in human retina. Ophthalmic Res. 1999, 31, 273–279. [Google Scholar] [CrossRef]
- Batliwala, S.; Xavier, C.; Liu, Y.; Wu, H.; Pang, I.H. Involvement of Nrf2 in ocular diseases. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Carneiro, L.S.F.; Fonseca, A.M.; Vieira-Coelho, M.A.; Mota, M.P.; Vasconcelos-Raposo, J. Effects of structured exercise and pharmacotherapy vs. pharmacotherapy for adults with depressive symptoms: A randomized clinical trial. J. Psychiatr. Res. 2015, 71, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Vinson, J. Oxidative stress in cataractogenesis. Pathophysiology 2006, 13, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Pescosolido, N.; Andrea, B.; Rossella, G.; Chiara, K.; Lenarduzzi, F. Age-related changes in the kinetics of human lenses: Prevention of the cataract. Int. J. Ophthalmol. 2016, 9, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Stefanson, A.L.; Bakovic, M. Dietary regulation of Keap1/Nrf2/ARE pathway: Focus on plant-derived compounds and trace minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef] [PubMed]
- Elanchezhian, R.; Palsamy, P.; Madson, C.J.; Mulhern, M.L.; Lynch, D.W.; Troia, A.M.; Usukura, J.; Shinohara, T. Low glucose under hypoxic conditions induces unfolded protein response and produces reactive oxygen species in lens epithelial cells. Cell Death Dis. 2012, 3, e301–e309. [Google Scholar] [CrossRef] [PubMed]
- Palsamy, P.; Ayaki, M.; Elanchezhian, R.; Shinohara, T. Promoter demethylation of Keap1 gene in human diabetic cataractous lenses. Biochem. Biophys. Res. Commun. 2012, 423, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, Y.; Huang, T. Human age-related cataracts: Epigenetic suppression of the nuclear factor erythroid 2-related factor 2-mediated antioxidant system. Mol. Med. Rep. 2015, 11, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Hao, J.L.; Xie, T.; Malik, T.H.; Lu, C.B.; Liu, C.; Shu, C.; Lu, C.W.; Zhou, D.D. Nrf2 as a target for prevention of age-related and diabetic cataracts by against oxidative stress. Aging Cell 2017, 16, 934–942. [Google Scholar] [CrossRef]
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef]
- Ibáñez, L.; Alcaraz, M.J.; Ferrándiz, M.L.; Cuadrado, A.; Brines, R.; Guede, D. Effects of Nrf2 Deficiency on Bone Microarchitecture in an Experimental Model of Osteoporosis. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef]
- Kim, J.H.; Singhal, V.; Biswal, S.; Thimmulappa, R.K.; DiGirolamo, D.J. Nrf2 is required for normal postnatal bone acquisition in mice. Bone Res. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Schultz, M.A.; Freeman, M.L.; Biswas, S. Loss of Nrf2 accelerates ionizing radiation-induced bone loss by upregulating RANKL. Free Radic. Biol. Med. 2012, 53, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-millan, M.; Brien, C.A.O.; Manolagas, S.C. Oxidative Stress Antagonizes Wnt Signaling in Osteoblast Precursors by Diverting β-Catenin from T Cell Factor- to Forkhead Box O-mediated Transcription. J. Biol. Chem. 2007, 282, 27298–27305. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Stephen, E.H.; Goutam, G.-C.; Nandini, G.-C. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 2011, 433, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, A.; Erdélyi, K.; Kovács, K.; Kovács, I.; Bai, P.; Rajnavölgyi, É.; Virág, L. Hydrogen peroxide-induced poly(ADP-ribosyl)ation regulates osteogenic differentiation-associated cell death. Free Radic. Biol. Med. 2012, 53, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. Bone 2014, 63, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Xu, A.H.; Yang, Y.; Li, J. Role of Nrf2 in bone metabolism. J. Biomed. Sci. 2015, 22, 101. [Google Scholar] [CrossRef] [PubMed]
- Lippross, S.; Beckmann, R.; Streubesand, N.; Ayub, F.; Tohidnezhad, M.; Campbell, G.; Kan, Y.W.; Horst, F.; Sönmez, T.T.; Varoga, D.; et al. Nrf2 Deficiency Impairs Fracture Healing in Mice. Calcif. Tissue Int. 2014, 95, 349–361. [Google Scholar] [CrossRef]
- Kanzaki, H.; Shinohara, F.; Kajiya, M.; Kodama, T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J. Biol. Chem. 2013, 288, 23009–23020. [Google Scholar] [CrossRef]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-d-aspartate Receptor-dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics Clin Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kashyap, M.P.; Tripathi, V.K. Neuroprotection Through Rapamycin-Induced Activation of Autophagy and PI3K/Akt1/mTOR/CREB Signaling Against Amyloid-β-Induced Oxidative Stress, Synaptic/Neurotransmission Dysfunction, and Neurodegeneration in Adult Rats. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. GSK3 β in Ethanol Neurotoxicity. Mol. Neurobiol. 2009, 40, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-isoe, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential Roles of the PI3 Kinase/Akt Pathway in Regulating Nrf2-Dependent Antioxidant Functions in the RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef] [PubMed]
- Mhillaj, E.; Catino, S.; Miceli, F.M.; Santangelo, R.; Trabace, L.; Cuomo, V.; Mancuso, C. Ferulic Acid Improves Cognitive Skills Through the Activation of the Heme Oxygenase System in the Rat. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Hong, B.; Fan, L.; Zhou, L.; Liu, Y.; Wu, Q.; Zhang, X.; Dong, M. Protective eff ect of puerarin against beta-amyloid-induced oxidative stress in neuronal cultures from rat hippocampus: Involvement of the GSK-3b/Nrf2 signaling pathway. Free Radic. Res. 2013, 47, 55–63. [Google Scholar] [CrossRef]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.; Goldsteins, G. Molecular and Cellular Neuroscience Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Raina, A.K.; Templeton, D.J.; Deak, J.C.; Perry, G.; Smith, M.A. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer’s disease. Redox Rep. 1999, 4, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Santa, K.; Decarli, C.; Johnson, J.A. NAD (P) H: Quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Alzheimer’s disease. Neurobiol. Aging 2000, 21, 525–531. [Google Scholar] [CrossRef]
- Santacruz, K.S.; Yazlovitskaya, E.; Collins, J.; Johnson, J.; Decarli, C. Regional NAD (P) H: Quinone oxidoreductase activity in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 63–69. [Google Scholar] [CrossRef]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F. Keap1 Is Localized in Neuronal and Glial Cytoplasmic Inclusions in Various Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef]
- Johnson, D.A.; Johnson, J.A. Author’s Accepted Manuscript Author’s Accepted Manuscript. Free Radic. Biol. Med. 2015. [Google Scholar] [CrossRef]
- Becker, I.-L. Role of the Transcription Factor Nrf2 in Parkinson’s Disease: New Insights. J. Alzheimers Dis. Park. 2017, 7. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.S.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 Is Necessary for Long Term Survival and Mitochondrial Function in Human Dopaminergic Neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokoshi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.H.; Yen, G.C. Differential expressions of antioxidant status in aging rats: The role of transcriptional factor Nrf2 and MAPK signaling pathway. Biogerontology 2007, 8, 71–80. [Google Scholar] [CrossRef]
- Van Muiswinkel, F.L.; de Vos, R.A.; Bol, J.G.; Andringa, G.; Jansen Steur, E.N.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Smith, M.A.; Richey, P.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Yoo, M.S.; Chun, H.S.; Son, J.J.; DeGiorgio, L.A.; Kim, D.J.; Peng, C.; Son, J.H. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: Correlation with the known pathology in Parkinson’s disease. Mol. Brain Res. 2003, 110, 76–84. [Google Scholar] [CrossRef]
- Cuadrado, A.; Moreno-Murciano, P.; Pedraza-Chaverri, J. The transcription factor Nrf2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Ther. Targets 2009, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Von Otter, M.; Landgren, S.; Nilsson, S.; Celojevic, D.; Bergström, P.; Håkansson, A.; Nissbrandt, H.; Drozdzik, M.; Bialecka, M.; Kurzawski, M.; et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson’s disease. BMC Med. Genet. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Von Otter, M.; Bergström, P.; Quattrone, A.; De Marco, E.V.; Annesi, G.; Söderkvist, P.; Wettinger, S.B.; Drozdzik, M.; Bialecka, M.; Nissbrandt, H.; et al. Genetic associations of Nrf2-encoding variants with Parkinson’s disease—A multicenter study. BMC Med. Genet. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, M.; Newman, J.R.B.; Shan, J.; Bentley, S.; Wood, S.A.; Silburn, P.A.; Mellick, G.D. Comprehensive assessment of genetic sequence variants in the antioxidant “master regulator” Nrf2 in idiopathic parkinson’s disease. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A.; Smith, L.M.; Coffey, M.P.; Aasly, J.O.; LeWitt, P.A. CSF Nrf2 and HSPA8 in Parkinson’s disease patients with and without LRRK2 gene mutations. J. Neural Transm. 2016, 123, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5. [Google Scholar] [CrossRef]
- Cookson, M.R. Parkinsonism Due to Mutations in PINK1,Parkin, and DJ-1 and Oxidative Stress and Mithochondrial Pathways. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Moscovitz, O.; Ben-Nissan, G.; Fainer, I.; Pollack, D.; Mizrachi, L.; Sharon, M. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Im, J.Y.; Lee, K.W.; Woo, J.M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef]
- Clements, C.M.; Mcnally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Tem, A.J.; Kalia, S.K.; Home, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; Garcıa, C.; Ruiz, J.F.; Grochot–Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different Susceptibility to the Parkinson’s Toxin MPTP in Mice Lacking the Redox Master Regulator Nrf2 or Its Target Gene Heme Oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [PubMed]
- Béraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoli, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein α-synuclein. J Neuroimmune Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-Specific Overexpression of Nrf2 Delays Motor Pathology and Synuclein Aggregation throughout the CNS in the Alpha-Synuclein Mutant (A53T) Mouse Model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef]
- He, Q.; Song, N.; Jia, F.; Xu, H.; Yu, X.; Xie, J.; Jiang, H. Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2013, 45, 1019–1030. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francisqueti-Ferron, F.V.; Ferron, A.J.T.; Garcia, J.L.; Silva, C.C.V.d.A.; Costa, M.R.; Gregolin, C.S.; Moreto, F.; Ferreira, A.L.A.; Minatel, I.O.; Correa, C.R. Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases. Int. J. Mol. Sci. 2019, 20, 3208. https://doi.org/10.3390/ijms20133208
Francisqueti-Ferron FV, Ferron AJT, Garcia JL, Silva CCVdA, Costa MR, Gregolin CS, Moreto F, Ferreira ALA, Minatel IO, Correa CR. Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases. International Journal of Molecular Sciences. 2019; 20(13):3208. https://doi.org/10.3390/ijms20133208
Chicago/Turabian StyleFrancisqueti-Ferron, Fabiane Valentini, Artur Junio Togneri Ferron, Jéssica Leite Garcia, Carol Cristina Vágula de Almeida Silva, Mariane Róvero Costa, Cristina Schmitt Gregolin, Fernando Moreto, Ana Lúcia A. Ferreira, Igor Otávio Minatel, and Camila Renata Correa. 2019. "Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases" International Journal of Molecular Sciences 20, no. 13: 3208. https://doi.org/10.3390/ijms20133208
APA StyleFrancisqueti-Ferron, F. V., Ferron, A. J. T., Garcia, J. L., Silva, C. C. V. d. A., Costa, M. R., Gregolin, C. S., Moreto, F., Ferreira, A. L. A., Minatel, I. O., & Correa, C. R. (2019). Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases. International Journal of Molecular Sciences, 20(13), 3208. https://doi.org/10.3390/ijms20133208