The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism


Abstract

1. Aerobic Glycolysis as Energetic Source in Cancer Cells
2. Molecular Network That Underlies Metabolic Reprogramming in Cancer
3. Role of Quercetin as an Anti-Cancer Agent—Molecular Implications
3.1. Effect in Cell Proliferation
3.2. Effect on Oxidative Stress
3.3. Effect in Autophagy and Apoptosis
3.4. Effect of Quercetin as Co-Adjuvant Agent in Anti-Cancer Therapy
3.5. Anti-Cancer Effect of Quercetin Derivatives
4. Anti-Cancer Effect of Quercetin by Targeting Key Molecular Factors in Anabolic Metabolism
5. Clinical Trials Addressing Anti-Cancer Effect of Quercetin
6. Conclusions
Abbreviations
| 2-DG | 2-Deoxyglucose |
| Akt | Protein Kinase B |
| Bax | bcl-2-like protein 4 |
| Bcl-2 | B-cell lymphoma 2 protein |
| Casp 3 | Caspase 3 |
| Casp 8 | Caspass 8 |
| Cox2 | Cyclooxygenase 2 |
| Cyt c | Cytochrome C |
| ERK | Extracellular signal–regulated kinases |
| GLUT1 | Glucose transporter 1 |
| GSK3 | Glycogen synthase kinase 3 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LC3-II | LC3-phosphatidylethanolamine conjugate |
| LDH | Lactate dehydrogenase |
| MAPK | Mitogen-activated protein kinase |
| mIMP | Mitochondrial inner membrane permeabilization |
| MKsi | Midkine silencer |
| MMP-2 | Matrix metalloproteinase-2 |
| MMP-9 | Matrix metalloproteinase-9 |
| mTOR | mammalian Target of Rapamycin |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| p21Cip1 | Cyclin-dependent kinase inhibitor 1 |
| p27Kip1 | Cyclin-dependent kinase inhibitor 1B |
| p38 | Mitogen activated protein kinase p38 |
| p53 | Tumor protein p53 |
| p70-S6K | Ribosomal protein S6 kinase beta-1 |
| PI3K | Phosphoinositide 3-kinase |
| PKM2 | Pyruvate kinase isozyme M2 |
| PTEN | phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase |
| ROS | Reactive oxygen species |
| Survivin | baculoviral inhibitor of apoptosis repeat-containing 5 |
| VEGF | Vascular endothelial growth factor |
| VEGF-R | Receptor of vascular endothelial growth factor |
| ΔΨm | mitochondrial membrane potential |
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). World Cancer Report 2014; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- GCO Estimated Number of Incident Cases from 2018 to 2040, all Cancers, Both Sexes, all Ages. Available online: http://gco.iarc.fr/tomorrow/home (accessed on 2 May 2019).
- Bray, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Zanetti, R.; Ferlay, J. CI5 XI: Cancer Incidence in Five Continents; Electronic Version; International Agency for Research on Cancer: Lyon, France, 2017; Volume XI. [Google Scholar]
- World Health Organization (WHO). Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 2 May 2019).
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Ghesquiere, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta. 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M. The Warburg Effect: Why and How Do Cancer Cells Activate Glycolysis in the Presence of Oxygen? Anti-Cancer Agents Med. Chem. 2008, 8, 305–312. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 7th ed.; WH Freeman: New York, NY, USA, 2017. [Google Scholar]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. The PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2015, 7, a011189. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. In Metabolism in Cancer; Cramer, T.A., Schmitt, C., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 39–72. [Google Scholar]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Marbaniang, C.; Kma, L. Dysregulation of Glucose Metabolism by Oncogenes and Tumor Suppressors in Cancer Cells. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 2377–2390. [Google Scholar] [PubMed]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Vegliante, R.; Ciriolo, M.R. Mitochondrial dysfunctions in cancer: Genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer Lett. 2015, 356, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Stavric, B. Quercetin in our diet: From potent mutagen to probable anticarcinogen. Clin. Biochem. 1994, 27, 245–248. [Google Scholar] [CrossRef]
- Perez-Jimenez, J.; Fezeu, L.; Touvier, M.; Arnault, N.; Manach, C.; Hercberg, S.; Galan, P.; Scalbert, A. Dietary intake of 337 polyphenols in French adults. Am. J. Clin. Nutr. 2011, 93, 1220–1228. [Google Scholar] [CrossRef]
- Ovaskainen, M.L.; Torronen, R.; Koponen, J.M.; Sinkko, H.; Hellstrom, J.; Reinivuo, H.; Mattila, P. Dietary intake and major food sources of polyphenols in Finnish adults. J. Nutr. 2008, 138, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, J.A.; Perez-Jimenez, J.; Neveu, V.; Medina-Remon, A.; M’Hiri, N.; Garcia-Lobato, P.; Manach, C.; Knox, C.; Eisner, R.; Wishart, D.S.; et al. Phenol-Explorer 3.0: A major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Datab. J. Biol. Datab. Curat. 2013, 2013, bat070. [Google Scholar] [CrossRef]
- Rothwell, J.A.; Urpi-Sarda, M.; Boto-Ordonez, M.; Knox, C.; Llorach, R.; Eisner, R.; Cruz, J.; Neveu, V.; Wishart, D.; Manach, C. Phenol-Explorer 2.0: A major update of the Phenol-Explorer database integrating data on polyphenol metabolism and pharmacokinetics in humans and experimental animals. Datab. J. Biol. Datab. Curat. 2012, 2012, bas031. [Google Scholar] [CrossRef]
- Neveu, V.; Perez-Jimenez, J.; Vos, F.; Crespy, V.; du Chaffaut, L.; Mennen, L.; Knox, C.; Eisner, R.; Cruz, J.; Wishart, D.; et al. Phenol-Explorer: An online comprehensive database on polyphenol contents in foods. Datab. J. Biol. Datab. Curat. 2010, 2010, bap024. [Google Scholar] [CrossRef]
- Hertog, M.G.; Hollman, P.C. Potential health effects of the dietary flavonol quercetin. Eur. J. Clin. Nutr. 1996, 50, 63–71. [Google Scholar]
- Carrasco-Pozo, C.; Tan, K.N.; Reyes-Farias, M.; De La Jara, N.; Ngo, S.T.; Garcia-Diaz, D.F.; Llanos, P.; Cires, M.J.; Borges, K. The deleterious effect of cholesterol and protection by quercetin on mitochondrial bioenergetics of pancreatic beta-cells, glycemic control and inflammation: In vitro and in vivo studies. Redox Biol. 2016, 9, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Cappello, A.R.; Frattaruolo, L.; Badolato, M.; Armentano, B.; Aiello, F. Quercetin and derivatives: Useful tools in inflammation and pain management. Future Med. Chem. 2017, 9, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Badolato, M.; Carullo, G.; Perri, M.; Cione, E.; Manetti, F.; Di Gioia, M.L.; Brizzi, A.; Caroleo, M.C.; Aiello, F. Quercetin/oleic acid-based G-protein-coupled receptor 40 ligands as new insulin secretion modulators. Fut. Med. Chem. 2017, 9, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Perri, M.; Manetti, F.; Aiello, F.; Caroleo, M.C.; Cione, E. Quercetin-3-oleoyl derivatives as new GPR40 agonists: Molecular docking studies and functional evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 1761–1764. [Google Scholar] [CrossRef] [PubMed]
- Saponara, S.; Sgaragli, G.; Fusi, F. Quercetin as a novel activator of L-type Ca(2+) channels in rat tail artery smooth muscle cells. Br. J. Pharmacol. 2002, 135, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Yarahmadi, A.; Khademi, F.; Mostafavi-Pour, Z.; Zal, F. In-Vitro Analysis of Glucose and Quercetin Effects on m-TOR and Nrf-2 Expression in HepG2 Cell Line (Diabetes and Cancer Connection). Nutr. Cancer 2018, 70, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Mukai, R.; Ishisaka, A. Quercetin and related polyphenols: New insights and implications for their bioactivity and bioavailability. Food Funct. 2015, 6, 1399–1417. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Bruno, R.S. Endogenous and exogenous mediators of quercetin bioavailability. J. Nutr. Biochem. 2015, 26, 201–210. [Google Scholar] [CrossRef]
- Jana, N.; Bretislav, G.; Pavel, S.; Pavla, U. Potential of the Flavonoid Quercetin to Prevent and Treat Cancer-Current Status of Research. Klin. Onkol. Cas. Ceske Slov. Onkol. Spol. 2018, 31, 184–190. [Google Scholar]
- Ren, M.X.; Deng, X.H.; Ai, F.; Yuan, G.Y.; Song, H.Y. Effect of quercetin on the proliferation of the human ovarian cancer cell line SKOV-3 in vitro. Exp. Ther. Med. 2015, 10, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; An, J.Y.; Kwon, Y.T.; Rhee, J.G.; Lee, Y.J. Effects of low dose quercetin: Cancer cell-specific inhibition of cell cycle progression. J. Cell. Biochem. 2009, 106, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.H.; Song, H.Y.; Zhou, Y.F.; Yuan, G.Y.; Zheng, F.J. Effects of quercetin on the proliferation of breast cancer cells and expression of survivin in vitro. Exp. Ther. Med. 2013, 6, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Amorim, R.; Pinheiro, C.; Miranda-Goncalves, V.; Pereira, H.; Moyer, M.P.; Preto, A.; Baltazar, F. Monocarboxylate transport inhibition potentiates the cytotoxic effect of 5-fluorouracil in colorectal cancer cells. Cancer Lett. 2015, 365, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Noori-Daloii, M.R.; Momeny, M.; Yousefi, M.; Shirazi, F.G.; Yaseri, M.; Motamed, N.; Kazemialiakbar, N.; Hashemi, S. Multifaceted preventive effects of single agent quercetin on a human prostate adenocarcinoma cell line (PC-3): Implications for nutritional transcriptomics and multi-target therapy. Med. Oncol. 2011, 28, 1395–1404. [Google Scholar] [CrossRef]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef]
- Mushtaq, M.; Gaza, H.V.; Kashuba, E.V. Role of the RB-Interacting Proteins in Stem Cell Biology. Adv. Cancer Res. 2016, 131, 133–157. [Google Scholar]
- Boly, R.; Gras, T.; Lamkami, T.; Guissou, P.; Serteyn, D.; Kiss, R.; Dubois, J. Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int. J. Oncol. 2011, 38, 833–842. [Google Scholar]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2016; Volume 81, pp. 163–175. [Google Scholar]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Maurya, A.K.; Vinayak, M. PI-103 and Quercetin Attenuate PI3K-AKT signaling Pathway in T- Cell Lymphoma Exposed to Hydrogen Peroxide. PLoS ONE 2016, 11, e0160686. [Google Scholar] [CrossRef] [PubMed]
- Hashemzaei, M.; Delarami Far, A.; Yari, A.; Heravi, R.E.; Tabrizian, K.; Taghdisi, S.M.; Sadegh, S.E.; Tsarouhas, K.; Kouretas, D.; Tzanakakis, G.; et al. Anticancer and apoptosisinducing effects of quercetin in vitro and in vivo. Oncol. Rep. 2017, 38, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, J.Y.; Ahn, K.S.; Cho, S.K. Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxid. Med. Cell. Longev. 2013, 2013, 596496. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gong, W.; Yang, Z.Y.; Zhou, X.S.; Gong, C.; Zhang, T.R.; Wei, X.; Ma, D.; Ye, F.; Gao, Q.L. Quercetin induces protective autophagy and apoptosis through ER stress via the p-STAT3/Bcl-2 axis in ovarian cancer. Apoptosis Int. J. Program. Cell Death 2017, 22, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Somasagara, R.R.; Hegde, M.; Nishana, M.; Tadi, S.K.; Srivastava, M.; Choudhary, B.; Raghavan, S.C. Quercetin, a Natural Flavonoid Interacts with DNA, Arrests Cell Cycle and Causes Tumor Regression by Activating Mitochondrial Pathway of Apoptosis. Sci. Rep. 2016, 6, 24049. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; et al. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1alpha-mediated signaling. Autophagy 2011, 7, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Voskas, D.; Ling, L.S.; Woodgett, J.R. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol. Rep. 2010, 2, B2–B82. [Google Scholar]
- Lei, C.S.; Hou, Y.C.; Pai, M.H.; Lin, M.T.; Yeh, S.L. Effects of quercetin combined with anticancer drugs on metastasis-associated factors of gastric cancer cells: In vitro and in vivo studies. J. Nutr. Biochem. 2018, 51, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Turkekul, K.; Dibirdik, I.; Doganlar, O.; Doganlar, Z.B.; Bilir, A.; Oktem, G. Midkine downregulation increases the efficacy of quercetin on prostate cancer stem cell survival and migration through PI3K/AKT and MAPK/ERK pathway. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 107, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Doganlar, Z.B.; Doganlar, O.; Turkekul, K.; Serttas, R. Inhibition of Midkine Suppresses Prostate Cancer CD133(+) Stem Cell Growth and Migration. Am. J. Med. Sci. 2017, 354, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Kadomatsu, K.; Goto, T.; Muramatsu, T. Combinational antitumor effect of siRNA against midkine and paclitaxel on growth of human prostate cancer xenografts. Cancer 2006, 107, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kang, N.J.; Heo, Y.S.; Rogozin, E.A.; Pugliese, A.; Hwang, M.K.; Bowden, G.T.; Bode, A.M.; Lee, H.J.; Dong, Z. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008, 68, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell Physiol. 2019, 27, 28334. [Google Scholar] [CrossRef] [PubMed]
- Dihal, A.A.; van der Woude, H.; Hendriksen, P.J.; Charif, H.; Dekker, L.J.; Ijsselstijn, L.; de Boer, V.C.; Alink, G.M.; Burgers, P.C.; Rietjens, I.M.; et al. Transcriptome and proteome profiling of colon mucosa from quercetin fed F344 rats point to tumor preventive mechanisms, increased mitochondrial fatty acid degradation and decreased glycolysis. Proteomics 2008, 8, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Sale, S.; Schmid, R.; Britton, R.G.; Brown, K.; Steward, W.P.; Gescher, A.J. Flavones as colorectal cancer chemopreventive agents--phenol-o-methylation enhances efficacy. Cancer Prev. Res. 2009, 2, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Hamidullah; Kumar, R.; Saini, K.S.; Kumar, A.; Kumar, S.; Ramakrishna, E.; Maurya, R.; Konwar, R.; Chattopadhyay, N. Quercetin-6-C-beta-D-glucopyranoside, natural analog of quercetin exhibits anti-prostate cancer activity by inhibiting Akt-mTOR pathway via aryl hydrocarbon receptor. Biochimie 2015, 119, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Csepregi, R.; Temesfői, V.; Sali, N.; Poór, M.; W Needs, P.; A Kroon, P.; Kőszegi, T. A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells. Int. J. Mol. Sci. 2018, 19, 2670. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Weissig, V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 17394–17421. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Targeting glucose metabolism: An emerging concept for anticancer therapy. Am. J. Clin. Oncol. 2011, 34, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Sturza, A.; Pavel, I.; Ancusa, S.; Danciu, C.; Dehelean, C.; Duicu, O.; Muntean, D. Quercetin exerts an inhibitory effect on cellular bioenergetics of the B164A5 murine melanoma cell line. Mol. Cell. Biochem. 2018, 447, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Maurya, A.K.; Vinayak, M. Quercetin regresses Dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr. Cancer 2015, 67, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.R.; Racker, E. Effects of quercetin and F1 inhibitor on mitochondrial ATPase and energy-linked reactions in submitochondrial particles. Biochim. Biophys. Acta 1974, 333, 180–186. [Google Scholar] [CrossRef]
- Suolinna, E.M.; Buchsbaum, R.N.; Racker, E. The effect of flavonoids on aerobic glycolysis and growth of tumor cells. Cancer Res. 1975, 35, 1865–1872. [Google Scholar] [PubMed]
- McCoy, G.D.; Racker, E. Protein synthesis in dextran sulfate-treated ascites tumor cells. Cancer Res. 1976, 36, 3346–3349. [Google Scholar] [PubMed]
- Racker, E.; Johnson, J.H.; Blackwell, M.T. The role of ATPase in glycolysis of Ehrlich ascites tumor cells. J. Biol. Chem. 1983, 258, 3702–3705. [Google Scholar]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef]
- Morris, M.E.; Felmlee, M.A. Overview of the proton-coupled MCT (SLC16A) family of transporters: Characterization, function and role in the transport of the drug of abuse gamma-hydroxybutyric acid. AAPS J. 2008, 10, 311–321. [Google Scholar] [CrossRef]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosén, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Takahashi, M.; Uramoto, H.; Nakayama, Y.; Oyama, T.; Wang, K.Y.; Sasaguri, Y.; Nishizawa, S.; Kohno, K. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011, 102, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Aslan, E.; Guler, C.; Adem, S. In vitro effects of some flavonoids and phenolic acids on human pyruvate kinase isoenzyme M2. J. Enzym. Inhib. Med. Chem. 2016, 31, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Reyes, A.M.; Velásquez, F.V.; Rivas, C.I.; Zhang, R.H.; Strobel, P.; Slebe, J.C.; Núñez-Alarcón, J.; Golde, D.W. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry 2001, 40, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Ma, J.; Han, J.; Li, M.; Shang, J. Pharmacokinetic comparison of quercetin, isoquercitrin, and quercetin-3-O-beta-D-glucuronide in rats by HPLC-MS. PeerJ 2019, 26, e6665. [Google Scholar] [CrossRef] [PubMed]
- de Blas, E.; Estan, M.C.; Del Carmen Gomez de Frutos, M.; Ramos, J.; Del Carmen Boyano-Adanez, M.; Aller, P. Selected polyphenols potentiate the apoptotic efficacy of glycolytic inhibitors in human acute myeloid leukemia cell lines. Regulation by protein kinase activities. Cancer Cell Int. 2016, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Xiong, L.; Liu, T.; Liu, X.; Chen, J.; Sun, S.Y.; Khuri, F.R.; Zong, Y.; Zhou, Q.; Zhou, W. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J. Biol. Chem. 2009, 284, 23225–23233. [Google Scholar] [CrossRef]
- Shang, H.S.; Lu, H.F.; Lee, C.H.; Chiang, H.S.; Chu, Y.L.; Chen, A.; Lin, Y.F.; Chung, J.G. Quercetin induced cell apoptosis and altered gene expression in AGS human gastric cancer cells. Environ. Toxicol. 2018, 33, 1168–1181. [Google Scholar] [CrossRef]
- Ma, Y.S.; Yao, C.N.; Liu, H.C.; Yu, F.S.; Lin, J.J.; Lu, K.W.; Liao, C.L.; Chueh, F.S.; Chung, J.G. Quercetin induced apoptosis of human oral cancer SAS cells through mitochondria and endoplasmic reticulum mediated signaling pathways. Oncol. Lett. 2018, 15, 9663–9672. [Google Scholar] [CrossRef]
- Mutlu Altundag, E.; Yilmaz, A.M.; Kocturk, S.; Taga, Y.; Yalcin, A.S. Synergistic Induction of Apoptosis by Quercetin and Curcumin in Chronic Myeloid Leukemia (K562) Cells. Nutr. Cancer 2018, 70, 97–108. [Google Scholar] [CrossRef]
- Carrasco-Torres, G.; Baltierrez-Hoyos, R.; Andrade-Jorge, E.; Villa-Trevino, S.; Trujillo-Ferrara, J.G.; Vasquez-Garzon, V.R. Cytotoxicity, Oxidative Stress, Cell Cycle Arrest, and Mitochondrial Apoptosis after Combined Treatment of Hepatocarcinoma Cells with Maleic Anhydride Derivatives and Quercetin. Oxidative Med. Cell. Longev. 2017, 2017, 2734976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Sulindac and Plant Compounds in Preventing Colon Cancer. 1996. Available online: https://ClinicalTrials.gov/show/NCT00003365 (accessed on 19 June 2019).
- Pilot Study Evaluating Broccoli Sprouts in Advanced Pancreatic Cancer POUDER Trial. 2013. Available online: https://ClinicalTrials.gov/show/NCT01879878 (accessed on 19 June 2019).
- Dietary Intervention in Follicular Lymphoma. 2007. Available online: https://ClinicalTrials.gov/show/NCT00455416 (accessed on 19 June 2019).
- Prostate Cancer Prevention Trial with Quercetin and Genistein. 2012. Available online: https://ClinicalTrials.gov/show/NCT01538316 (accessed on 19 June 2019).
- Lozanovski, V.J.; Houben, P.; Hinz, U.; Hackert, T.; Herr, I.; Schemmer, P. Pilot study evaluating broccoli sprouts in advanced pancreatic cancer (POUDER trial) - study protocol for a randomized controlled trial. Trials 2014, 15, 204. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-Cancer Effects of Green Tea Polyphenols Against Prostate Cancer. Molecules 2019, 24, 193. [Google Scholar] [CrossRef]
- Wang, P.; Heber, D.; Henning, S.M. Quercetin increased the antiproliferative activity of green tea polyphenol (-)-epigallocatechin gallate in prostate cancer cells. Nutr. Cancer 2012, 64, 580–587. [Google Scholar] [CrossRef]
- Kale, A.; Gawande, S.; Kotwal, S.; Netke, S.; Roomi, W.; Ivanov, V.; Niedzwiecki, A.; Rath, M. Studies on the effects of oral administration of nutrient mixture, quercetin and red onions on the bioavailability of epigallocatechin gallate from green tea extract. Phytother. Res. 2010, 24, S48–S55. [Google Scholar] [CrossRef]
- Wang, P.; Heber, D.; Henning, S.M. Quercetin increased bioavailability and decreased methylation of green tea polyphenols in vitro and in vivo. Food Funct. 2012, 3, 635–642. [Google Scholar] [CrossRef]
- Henning, S.M.; Aronson, W.J.; Wang, P.; Wang, J.; Lee, R.-P.; Grojean, E.M.; Ly, A.; Hsu, M.; Heber, D.; Li, Z. Combination of Quercetin and Green Tea Extract Increased Plasma Epicatechingallate and Decreased Urine Epigallocatechin and Epicatechin Concentrations. FASEB J. 2017, 31, 148.8. [Google Scholar]
- Effect of Quercetin on Green Tea Polyphenol Uptake in Prostate Tissue from Patients with Prostate Cancer Undergoing Surgery. 2014. Available online: https://ClinicalTrials.gov/show/NCT01912820 (accessed on 19 June 2019).
- Quercetin Chemoprevention for Squamous Cell Carcinoma in Patients with Fanconi Anemia. 2018. Available online: https://ClinicalTrials.gov/show/NCT03476330 (accessed on 19 June 2019).
- Thomas, P.; Holland, N.; Bolognesi, C.; Kirsch-Volders, M.; Bonassi, S.; Zeiger, E.; Knasmueller, S.; Fenech, M. Buccal micronucleus cytome assay. Nat. Protoc. 2009, 4, 825–837. [Google Scholar] [CrossRef]
- Suolinna, E.M.; Lang, D.R.; Racker, E. Quercetin, an artificial regulator of the high aerobic glycolysis of tumor cells. J. Natl. Cancer Inst. 1974, 53, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Weidemann, M.J.; Ferber, E. Preferential inhibition by quercetin of mitogen-stimulated thymocyte glucose transport. J. Natl. Cancer Inst. 1979, 62, 1243–1246. [Google Scholar]
- Belt, J.A.; Thomas, J.A.; Buchsbaum, R.N.; Racker, E. Inhibition of lactate transport and glycolysis in Ehrlich ascites tumor cells by bioflavonoids. Biochemistry 1979, 18, 3506–3511. [Google Scholar] [CrossRef] [PubMed]

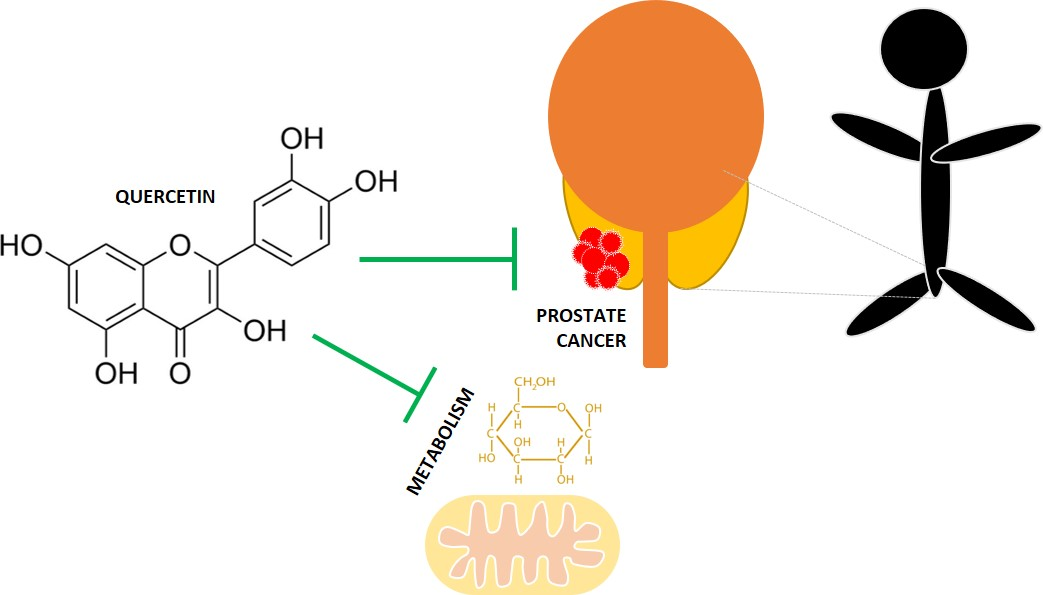{kind=link}
| Model | QUE Concentration | Anti-Cancer Effects | Reference |
|---|---|---|---|
| AGS cell line | 6.25 to 100 μM | Reduced cell viability in a concentration-dependent manner; at dose 50 µM inhibited 50% growth. When added to SN-38, it improved the anti-proliferation effect of this compound, and increased apoptosis, acting synergistically with SN-38 in the modulation of GSK-3β/β-catenin signaling. | [76] |
| BALB/c nude mice injected with AGS cells | 20 mg/kg BW | QUE alone (three times per week) or in combination with irinotecan (10 mg/kg once per week), promoted a significant reduction of tumor size at day 28, and also reduced tumor VEGF-R and VEGF-A levels, protein levels, and reduced COX-2 gene expression. This combination also decreased the TEM population. | [76] |
| MCF-7 and MDA-MB-231 cell lines | 0 to 100 μM | Decreased viability (IC50 = 30 μM), increased autophagy, suppressed migration rate and reduced MMP-2, MMP-9 and VEGF protein levels. Also, suppressed glucose uptake, lactate production, and expression of PKM2, LDHA, and GLUT1. Similarly, QUE suppressed activation of AKT, mTOR, and p70-S6K. | [64] |
| Mice injected with MCF-7 cells | 50 mg/kg BW | Inhibited tumor metastasis and progression of breast cancer. It also decreased VEGF, PKM2, and p-AKT levels in tumor tissue. | [64] |
| PC3 cells | 1.78 to 100 μM | Inhibited survival in a dose- and time-dependent manner. At a concentration of 40 μM, QUE increased Cyt c, casp 3, casp 8, Bax, Bcl-2, p21Cip1, p27Kip1, and p53. QUE improved apoptotic effect of MKsi, increased casp 3 and decreased Survivin gene expression. QUE promoted the arrest in the G1 phase cells and decreased cells in the S-phase. QUE decreased the phosphorylation of PI3K, Akt, and ERK1/2. QUE decreased p38, NFκB, and Survivin protein levels and increased the PTEN expression. Combined with MKsi, QUE had a greater effect over ERK1/2, p38, NFκB, and Survivin. | [77] |
| B164A5 murine melanoma cells | 150 μM at 72 h | Reduced OCR and ECAR. | [90] |
| BC3, BCBL1, and BC1 PEL cell lines. | 12 to 100 μM | QUE for 24 h reduced cell survival and growth in a dose-dependent manner, without affecting normal B lymphocytes. QUE 50 μM increased apoptosis rate, increasing the G1 cell phase, PARP cleavage, and nuclear fragmentation/condensation. At this concentration, QUE also inhibited mTOR and Aktser473 and promoted degradation of β-catenin. | [62] |
| MCF-7, MDA-MB-231, HBL100 and BT549 breast cancer cells, and OVCAR5, TOV112D, OVCAR3, CAOV3 ovarian cancer cells. | 0.6 to 300 μΜ | Reduced cell proliferation concentration-dependently. | [58] |
| HBL100 cells | 50 μΜ for 24 h | Increased intracellular accumulation of glucose and promoted lactate depletion into the culture media. | [58] |
| MCF-7 cells | 300 μΜ for 48 h | Increased apoptosis by 25%. | [58] |
| HCT-15 and RKO cells | 0 to 200µM | Inhibited cell proliferation, viability, and promoted apoptosis in a concentration dependent manner in cancer cells, but not in normal cells. | [61] |
| HCT-15 cells | 142.7 µM (IC50) | Reduced glucose consumption and lactate production after 4 h incubation. QUE increased sensitization to 5-FU, improving its effects in glucose metabolism inhibition. | [61] |
| RKO cells | 121.9 µM (IC50) | QUE increased sensitization to 5-FU, improving its effects in glucose metabolism inhibition. | [61] |
| DL mice | 25 to 75 mg/kg BW | Decreased cell viability, mRNA expression and activity of LDH-A, in a dose-dependent manner, without generating liver toxicity. QUE downregulated p85a phosphorylation and Akt gene/protein expression and up-regulated mRNA expression of p53. | [91] |
| Ehrlich ascites tumor cells | 26.5 µM | Inhibited lactate production by 78% and of Na+-K+-ATPase by 85%. | [93] |
| Ehrlich ascites tumor cells | 13.25 to 66.17 µM | At a concentration of 26.5 µM, and after 10 min of treatment, QUE caused 50% inhibition in Na+-K+-ATPase activity. QUE inhibited aerobic glycolysis and oxidative phosphorylation in a concentration-dependent manner. | [92,126] |
| Ascites tumor cells | 33.09 µM | Inhibited glycolysis and protein synthesis. | [94] |
| Rat thymocytes | 25 µM | Prevented glucose uptake induced by mitogenic stimulus. | [127] |
| Ascites tumor cells | 0.1µg/mg protein | Inhibited lactate efflux by 50%, increasing internal lactate concentration and decreasing intracellular pH. | [128] |
| HL60 cells | 5 to 40 μM | QUE, and in combination with 2-DG, induced caspase-dependent late apoptosis, decrease of mitochondrial membrane potential and induction of mIMP, and attenuated Akt and rpS6 phosphorylation. Co-treatment with PI3K/Akt phosphorylation inhibitors increases the apoptosis rate at low concentrations of QUE (10μM) and 2-DG (2 mM). | [103] |
| Recombinant human PKM2 enzyme | 9.24 µM | Inhibited PKM2 activity by 50%. | [100] |
| Clinical Study Title | Description | Dose, Via and Frequency of Administration | Benefits | Limitations | Ref |
|---|---|---|---|---|---|
| Sulindac and plant compounds in preventing colon cancer | Study the effectiveness of QUE among other compounds in preventing colon cancer. Estimated enrollment: Not specified Allocation: Randomized Primary Purpose: Prevention | Orally administered. 1 of 3 doses twice daily. For 6–10 weeks. | Determination of the lowest effective dose of QUE in modulating biomarkers of colon epithelial cell turnover, as an indication of colon cancer prevention. | No results published, although study completion date was 2006. | [113] |
| Pilot study evaluating broccoli sprouts in advanced pancreatic cancer | Administration of freeze-dried broccoli sprouts rich in QUE and sulforaphane in patients with advanced pancreatic ductal adenocarcinoma Estimated enrollment: 40 participants Allocation: Randomized Intervention Model: Parallel assignment | Orally administered. Capsules with broccoli sprout grain (90 mg sulforaphane daily + QUE-dose not specified). For 12 months. | Evaluation of cancer progress or regress in supplementation with capsules rich in QUE. | This study only declared sulforaphane concentration, but QUE content in the sprout was not specified. No results published, although study completion date was 2015. | [114] |
| Dietary Intervention in follicular lymphoma (Phase 2) | Assessment of the ability of grape juice (rich in QUE), among several dietary factors, to induce apoptosis, inhibit cell proliferation and modulate tumor cell infiltrate in vivo. Estimated enrollment: 45 participants Allocation: Non-Randomized Intervention Model: Single group assignment | Orally administered. Merlot grape juice 100%, 660 mL /495 mL every second day. For 16 weeks. | Determination of apoptosis of tumor cells as parameter of intervention efficacy. | The QUE content of the juice is unknown. The study completion date was 2009, however no results have been published yet. | [115] |
| Prostate cancer prevention with QUE and genistein | Evaluation of the effect of QUE or genistein supplementation, in comparison with placebo, against a PSA (prostate-specific antigen) increase. Estimated enrollment: 60 participants Allocation: Randomized Intervention Model: Crossover assignment | Orally administered. 500 mg QUE + vitamin C + folic acid + vitamin B3 daily. For 6 months. | Determination of QUE effect in PSA levels | This study evaluated a QUE supplement combined with other compounds that could act synergically or impact negatively over QUE effect, inducing side effects. | [116] |
| Effect of QUE on green tea polyphenol uptake in prostate tissue from patients with prostate cancer undergoing surgery (Phase 1) | Evaluation of the ability of QUE to enhance the uptake of green tea polyphenols in the prostate tissue of men taking green tea extract and undergoing radical prostatectomy. Evaluation of side effects of QUE in combination with green tea. Estimated enrollment: 31 participants Allocation: Randomized Intervention Model: Parallel assignment Primary Purpose: Prevention | Orally administered. Green tea polyphenol + QUE twice daily. For 3–6 weeks (before undergoing prostatectomy). | Determination of epigallocatechin gallate, epicatechin gallate and QUE concentration, and their methylated metabolites in prostate tissue and plasma. Determination of the extract and QUE on reducing the enzyme activity and protein and gene expression of catechol-O-methyltransferase (COMT), deoxyribonucleic methyltransferase 1 (DNMT1), and multidrug resistance transport protein 1 (MRP1) in prostate tissue. | The concentration of the extract and QUE used is not indicated. | [123] |
| Quercetin chemoprevention for squamous cell carcinoma in patients with fanconi anemia (FA) (Phase 2) | Efficacy of QUE in reducing buccal squamous cell carcinoma. Estimated enrollment: 55 participants Intervention Model: Single group assignment Primary Purpose: Prevention | Orally administered. Twice daily at an adjusted dose based on weight for a maximum total daily dose of 4000 mg/day. For 24 months | Evaluation of the efficacy of QUE in reducing buccal micronuclei and the need for potentially lethal treatment with chemotherapy and/or radiation therapy. | The primary outcome of this study is the reduction of buccal micronuclei in 45 post-hematopoietic cell transplantation (HCT) patients with FA. This is compared to only 10 patients FA patients without a history of HCT. | [124] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. https://doi.org/10.3390/ijms20133177
Reyes-Farias M, Carrasco-Pozo C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. International Journal of Molecular Sciences. 2019; 20(13):3177. https://doi.org/10.3390/ijms20133177
Chicago/Turabian StyleReyes-Farias, Marjorie, and Catalina Carrasco-Pozo. 2019. "The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism" International Journal of Molecular Sciences 20, no. 13: 3177. https://doi.org/10.3390/ijms20133177
APA StyleReyes-Farias, M., & Carrasco-Pozo, C. (2019). The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. International Journal of Molecular Sciences, 20(13), 3177. https://doi.org/10.3390/ijms20133177