Carbon Monoxide Releasing Molecule-2-Upregulated ROS-Dependent Heme Oxygenase-1 Axis Suppresses Lipopolysaccharide-Induced Airway Inflammation



Abstract
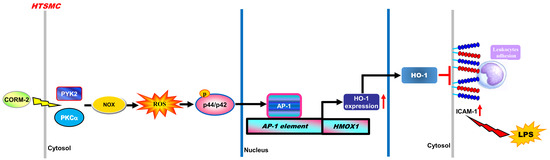
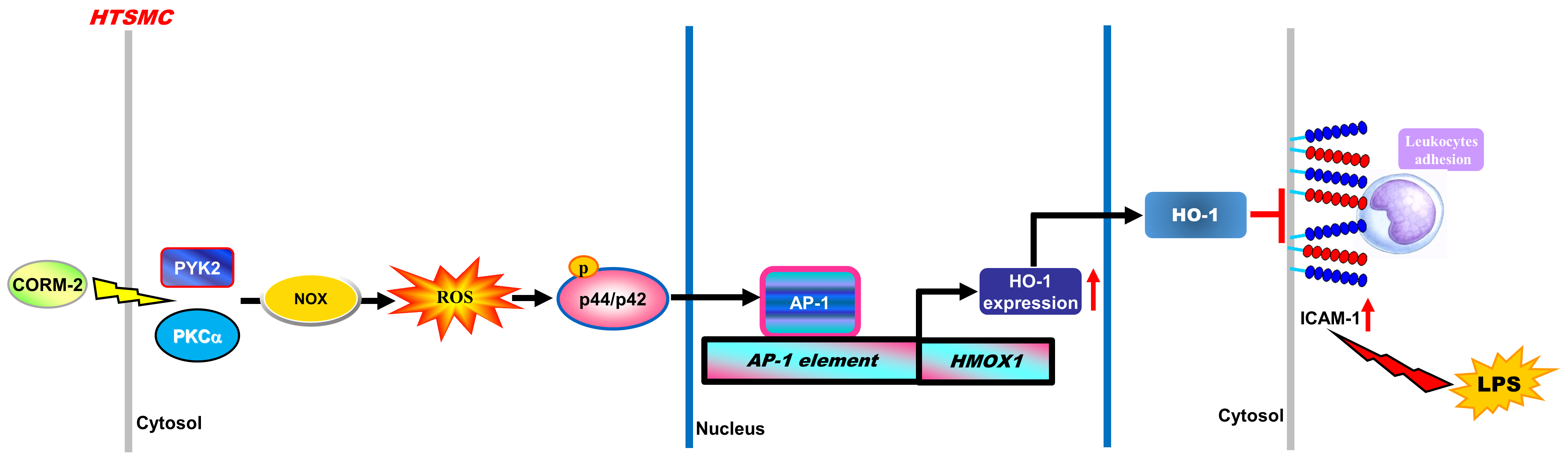{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
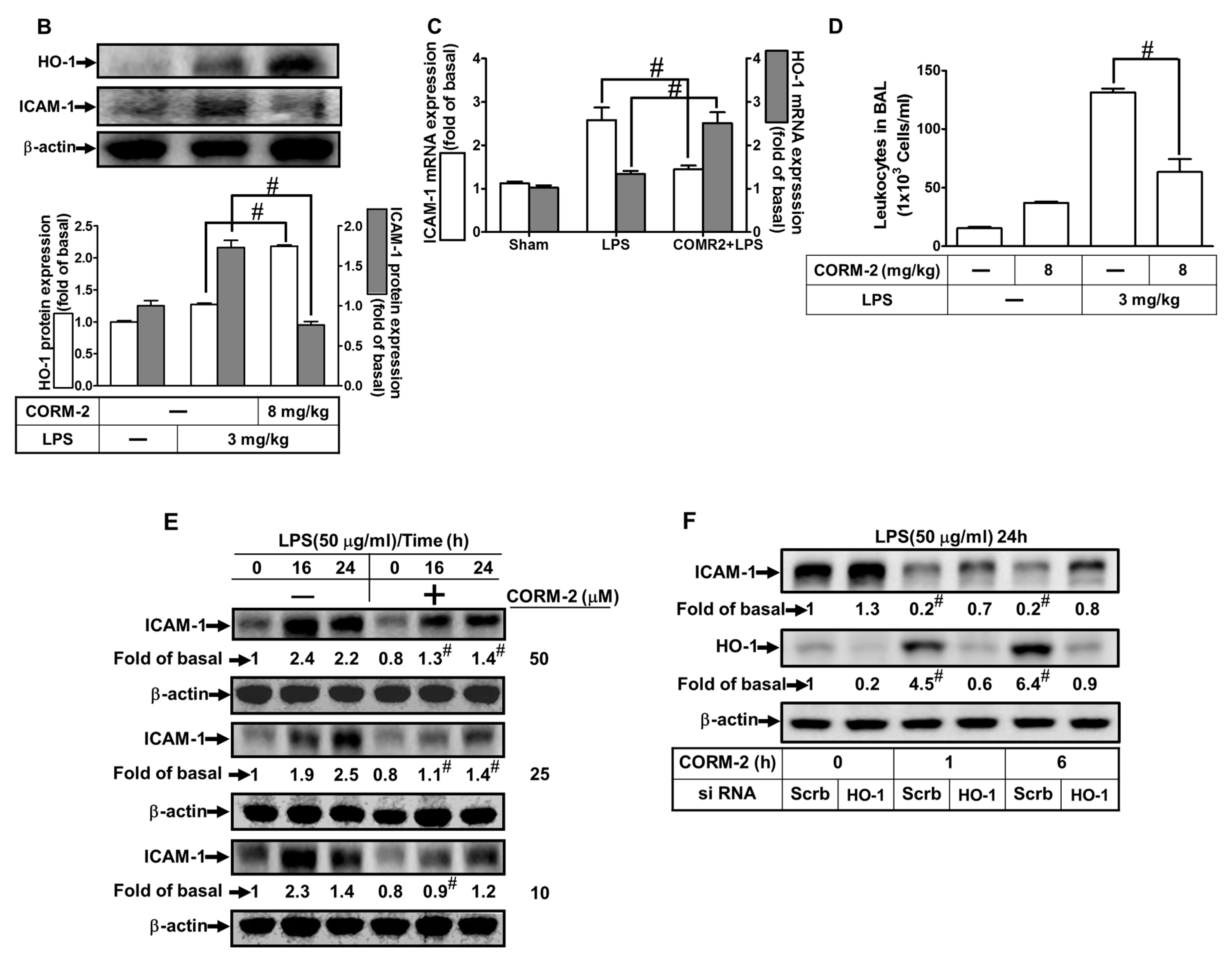
2.1. CORM-2 Inhibits LPS-Induced Lung Inflammation in Mice
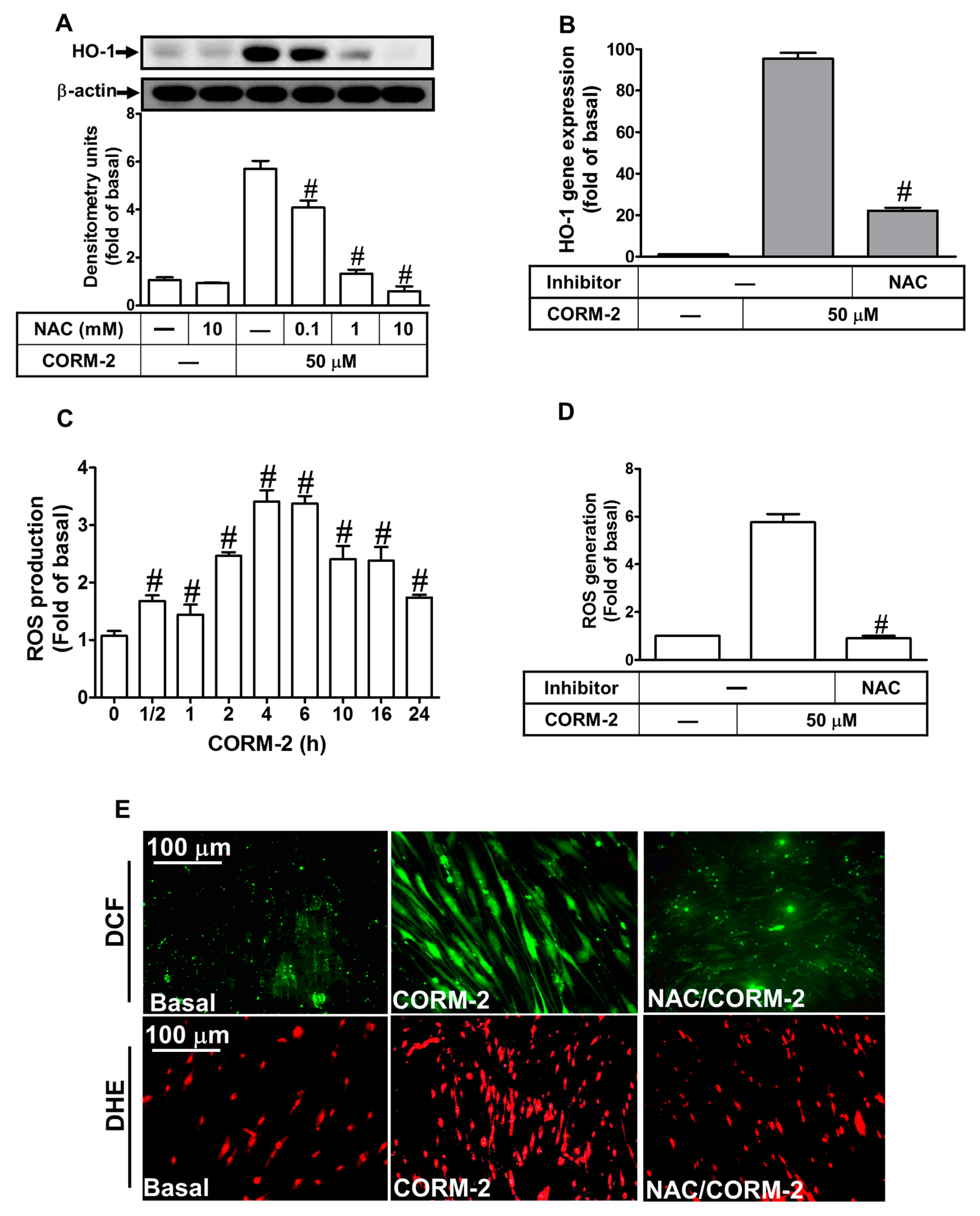
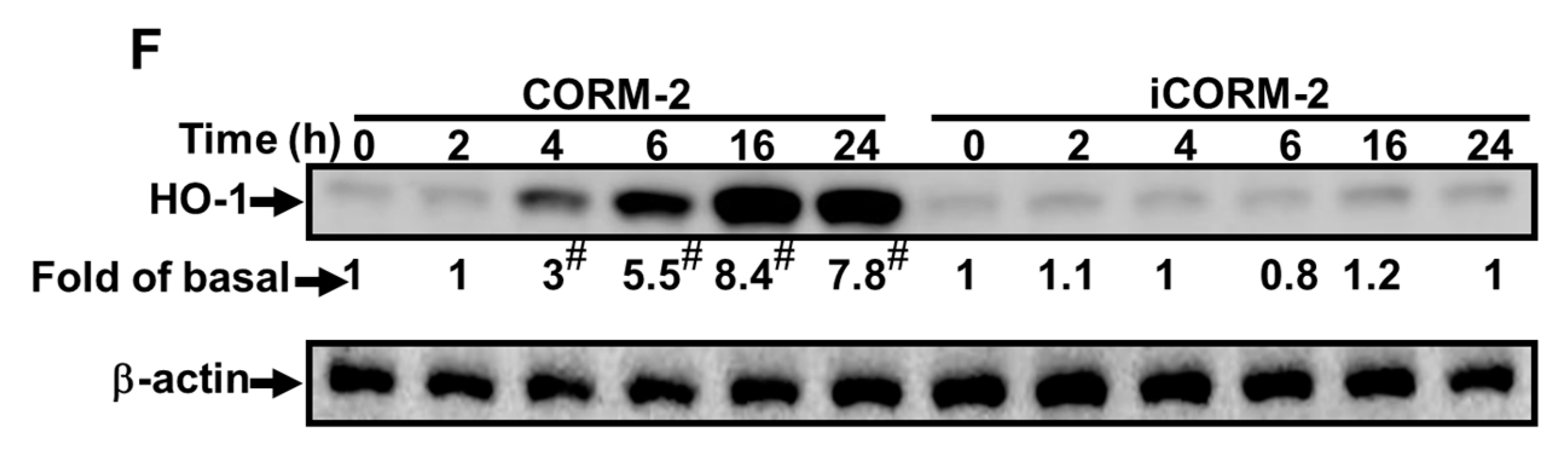
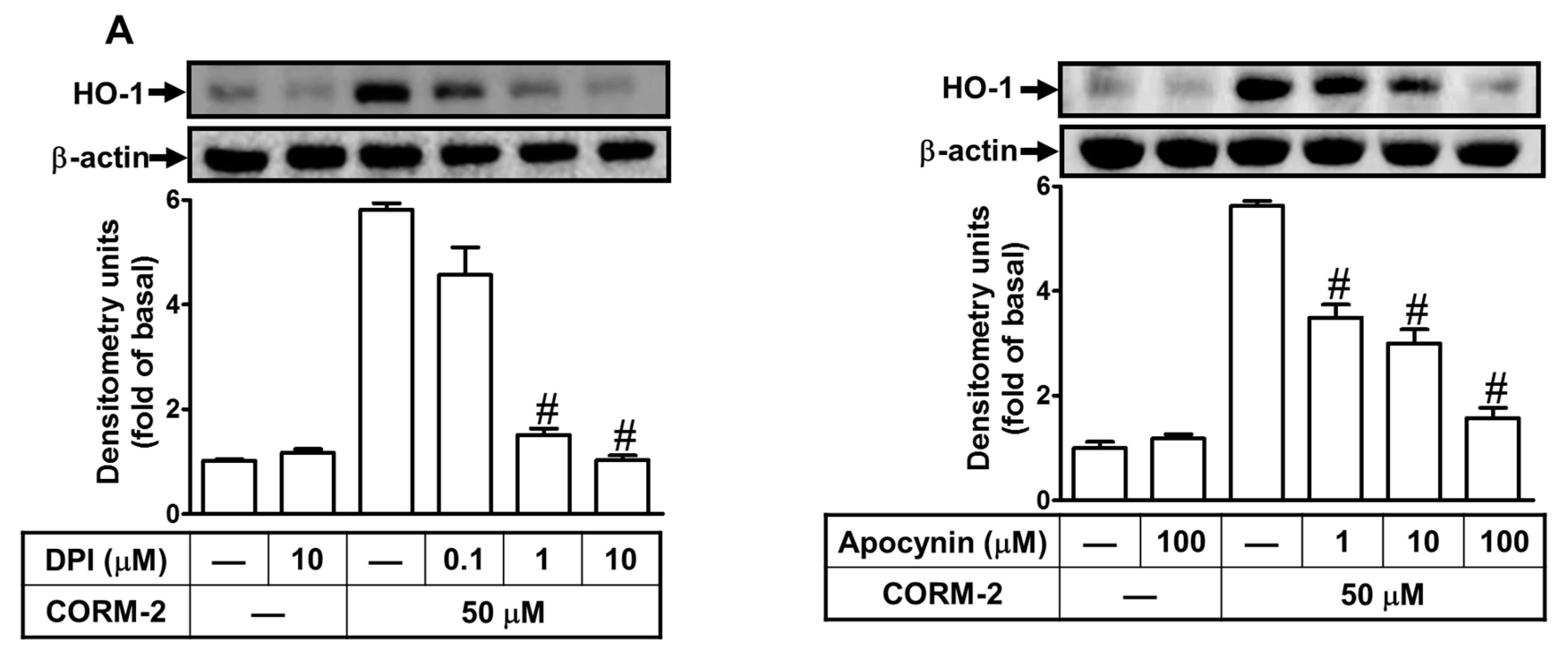
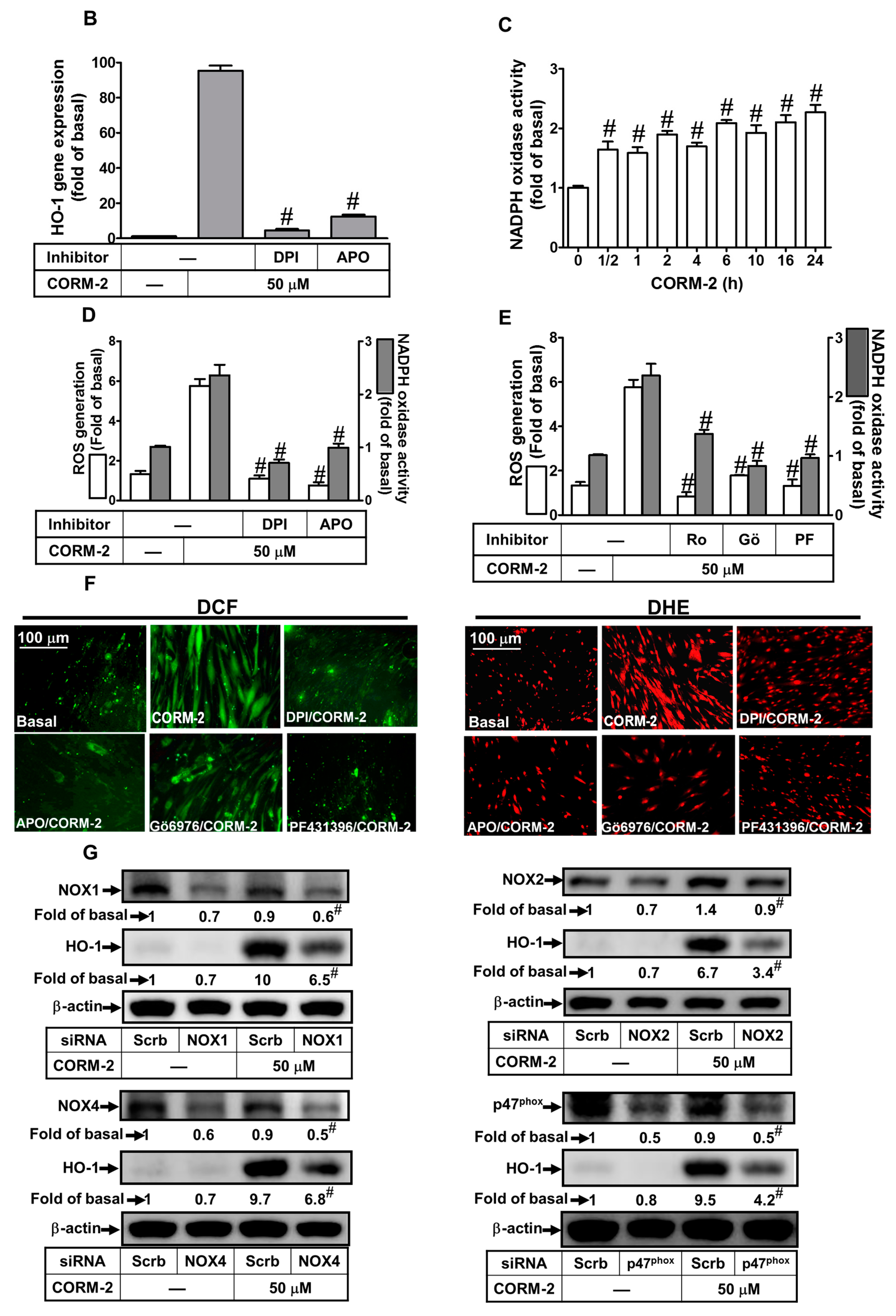
2.2. ROS Participate in CORM-2-Induced HO-1 Expression
2.3. Nox-Derived ROS Generation Contributes to CORM-2-Induced HO-1 Expression
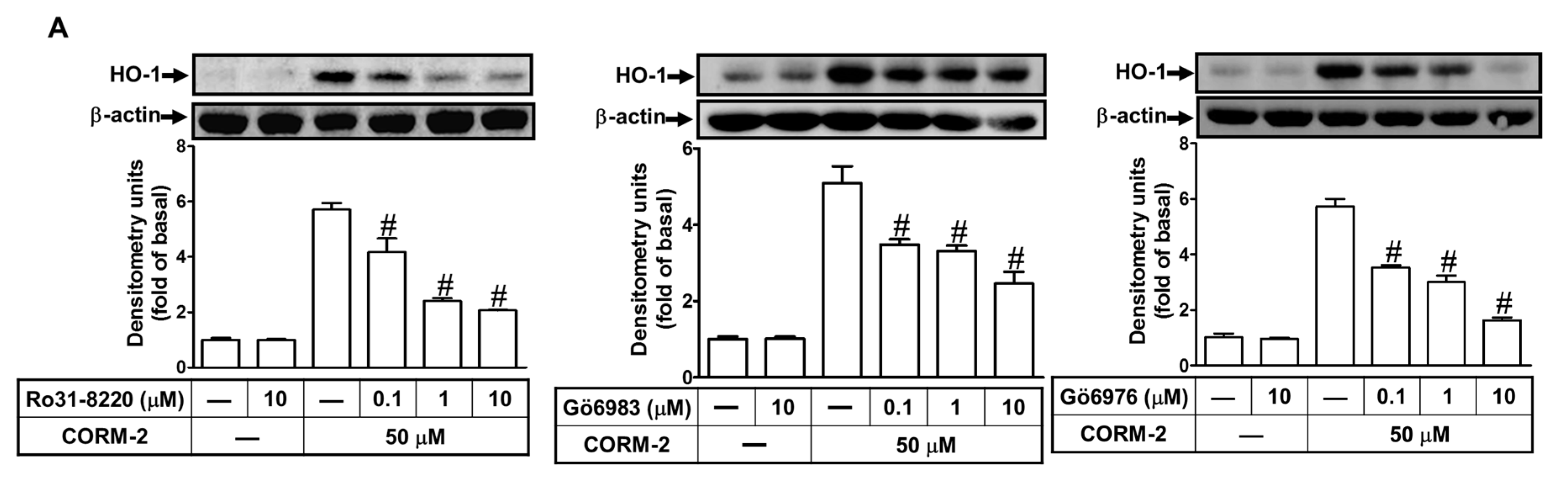
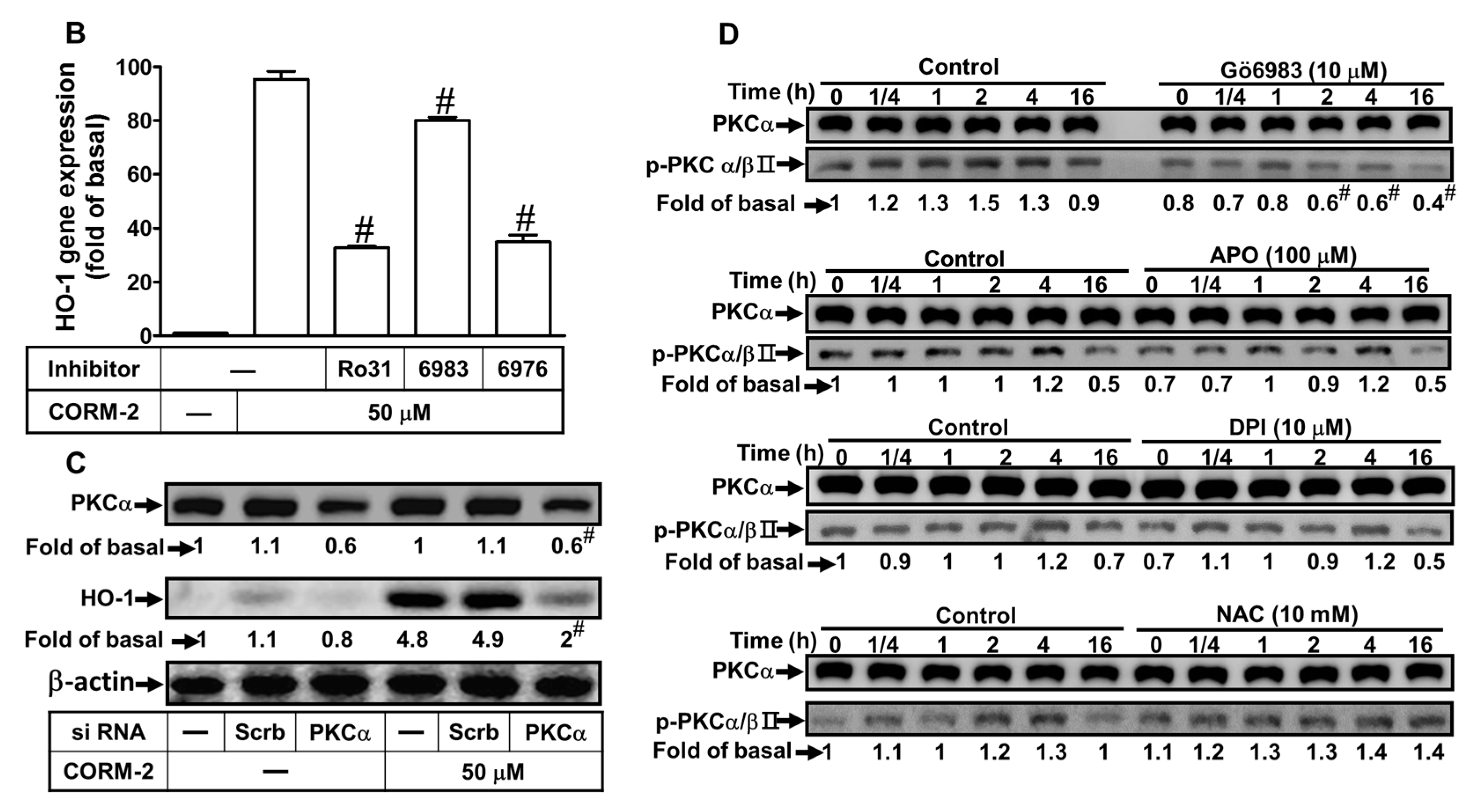
2.4. CORM-2 Induces HO-1 Expression via PKCα
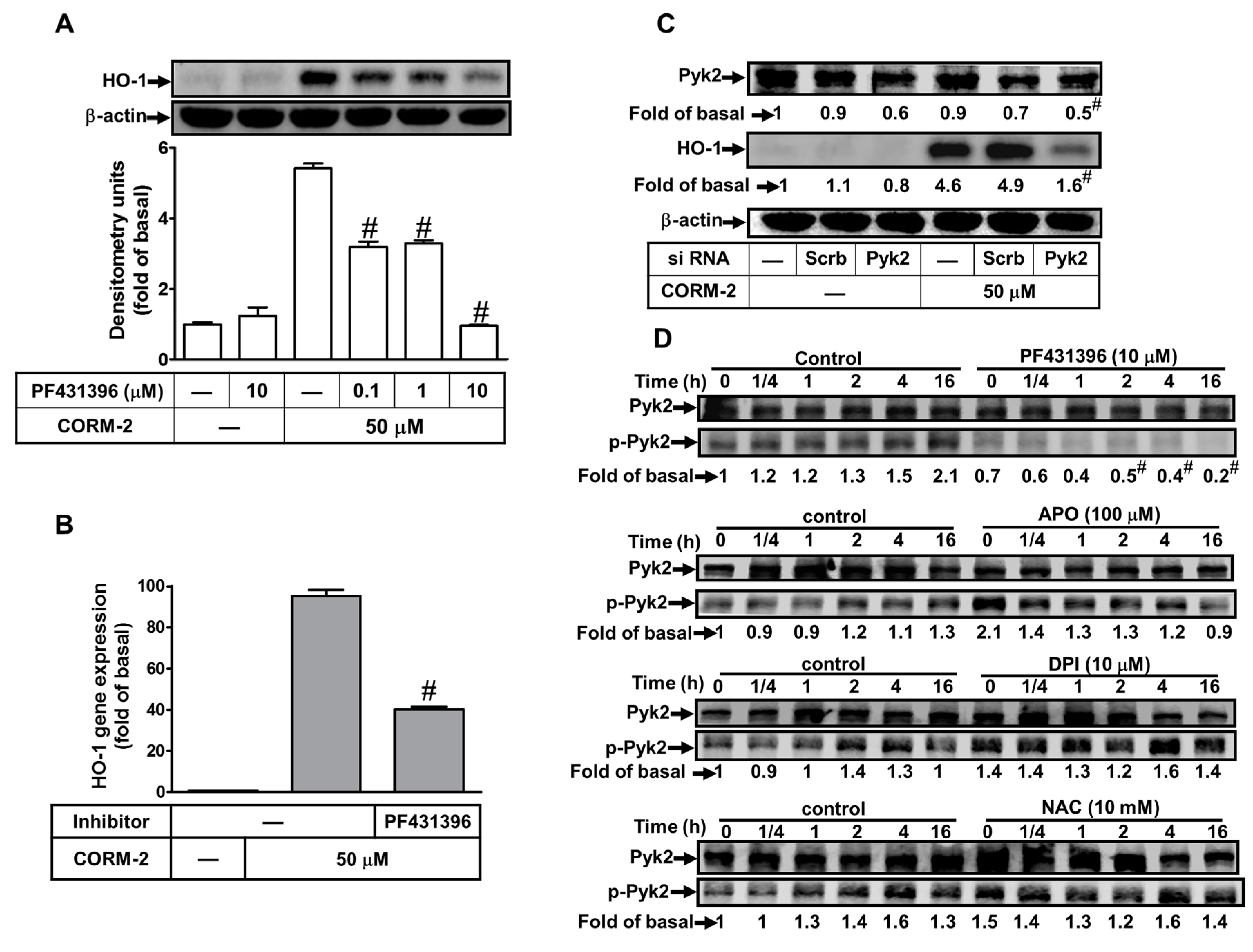
2.5. Involvement of Pyk2 in CORM-2-Induced HO-1 Expression
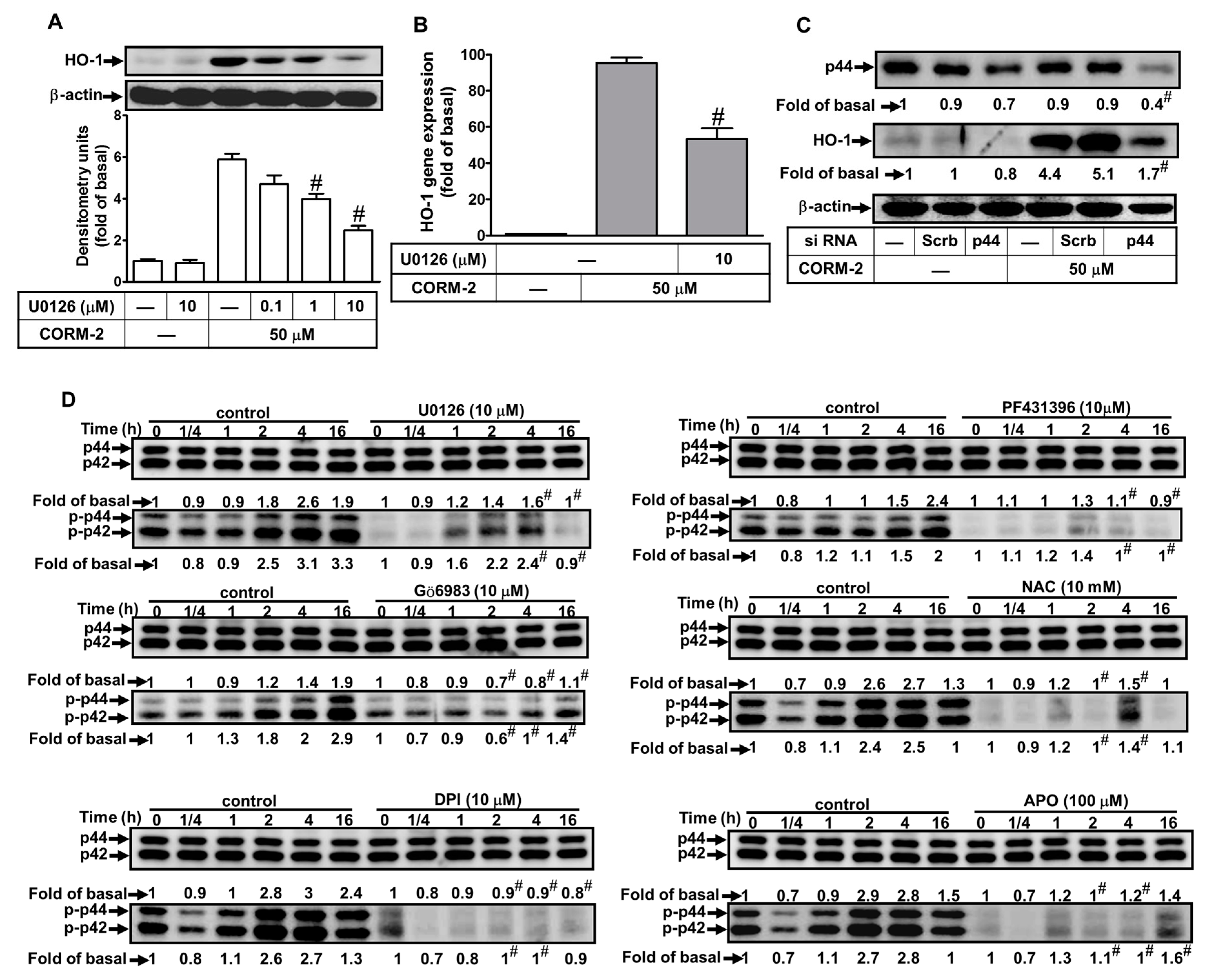
2.6. Involvement of ERK1/2 in CORM-2-Induced HO-1 Expression
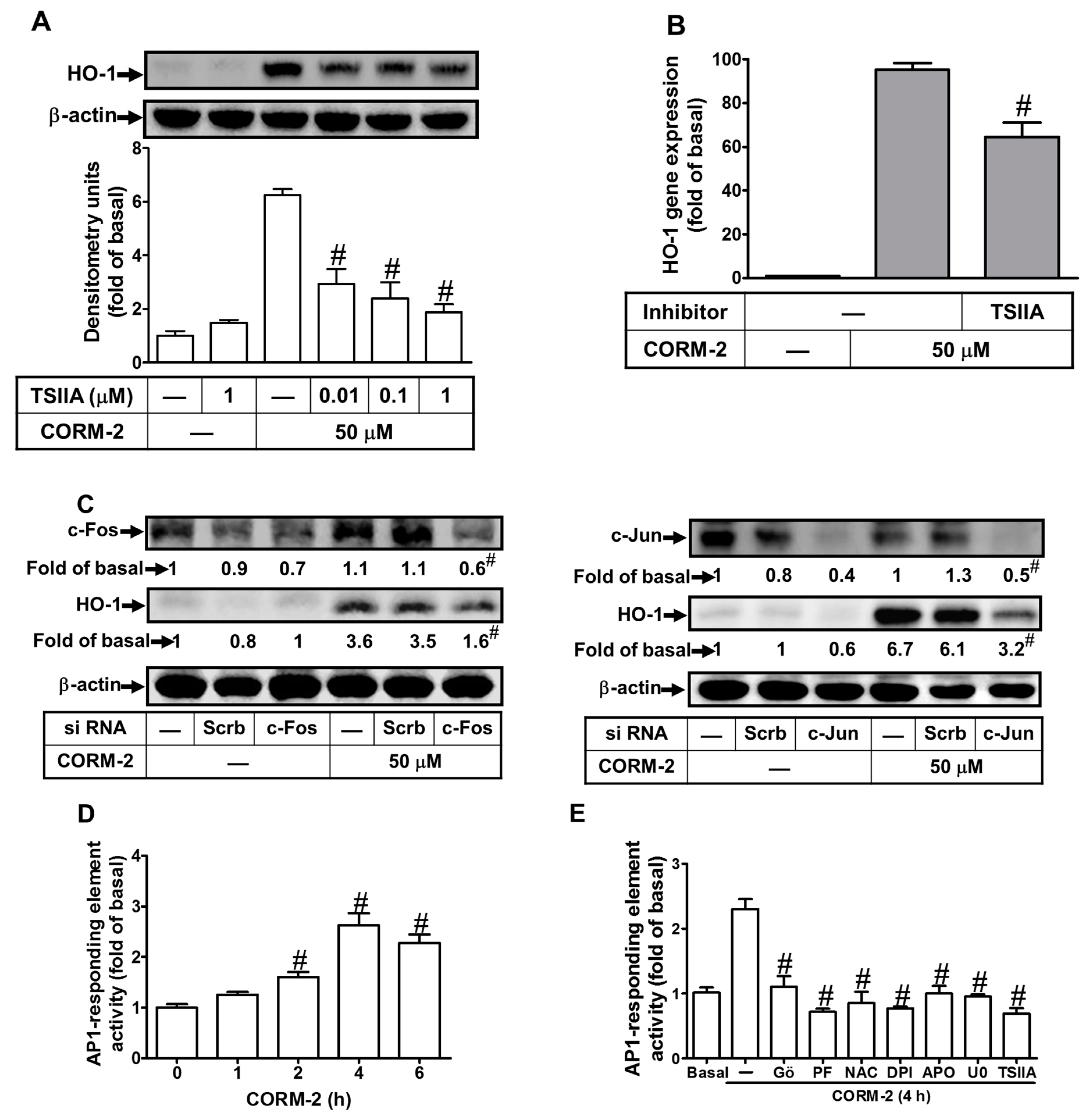
2.7. Induction of c-Fos and c-Jun/AP-1 is Required for CORM-2-Induced HO-1 Expression
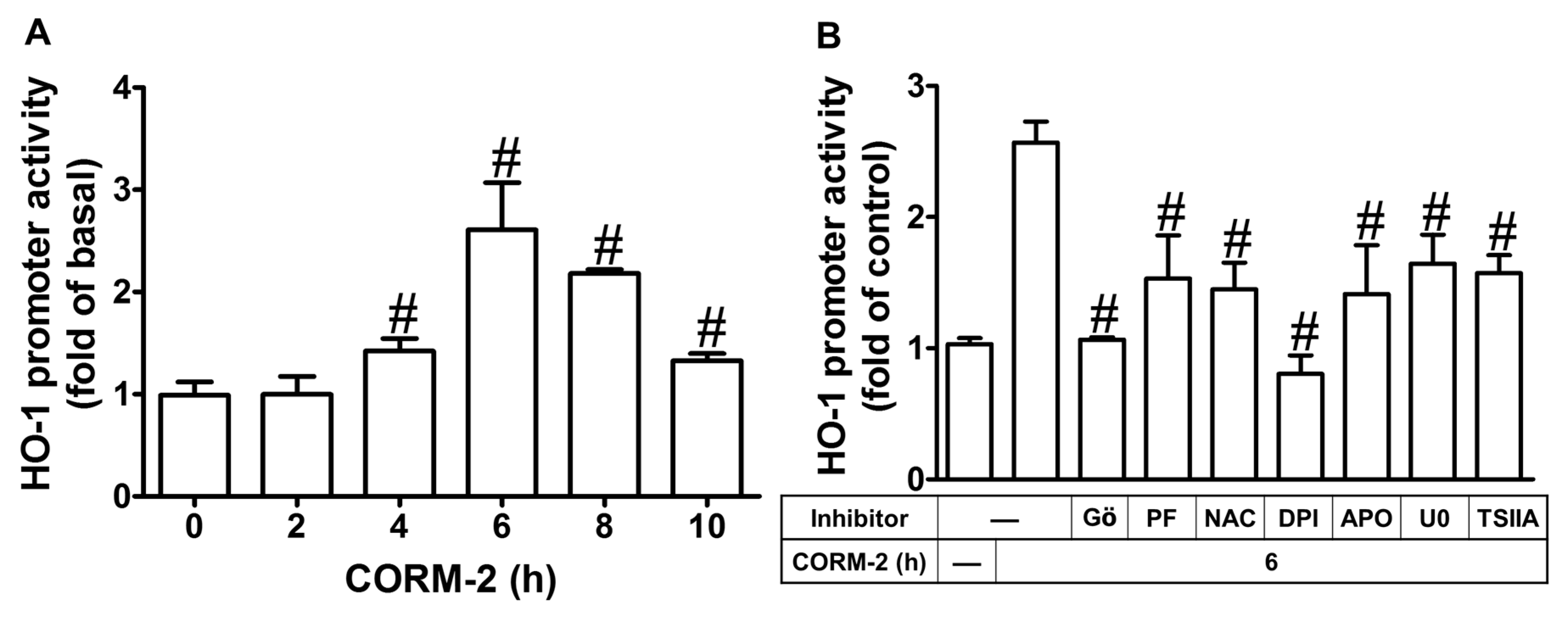
2.8. CORM-2 Induces HO-1 Expression via Its Promoter Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Animal Care and Experimental Procedures
4.3. Cell Culture and Treatment
4.4. Preparation of Cell Extracts and Western Blot Analysis
4.5. Total RNA Extraction and Real Time-Quantitative PCR Analysis
4.6. Plasmid Construction, Transfection, and Luciferase Reporter Gene Assays
4.7. Transient Transfection with siRNAs
4.8. Measurement of Intracellular ROS Generation
4.9. Determination of NADPH Oxidase Activity by Chemiluminescence Assay
4.10. Statistical Analysis of Data
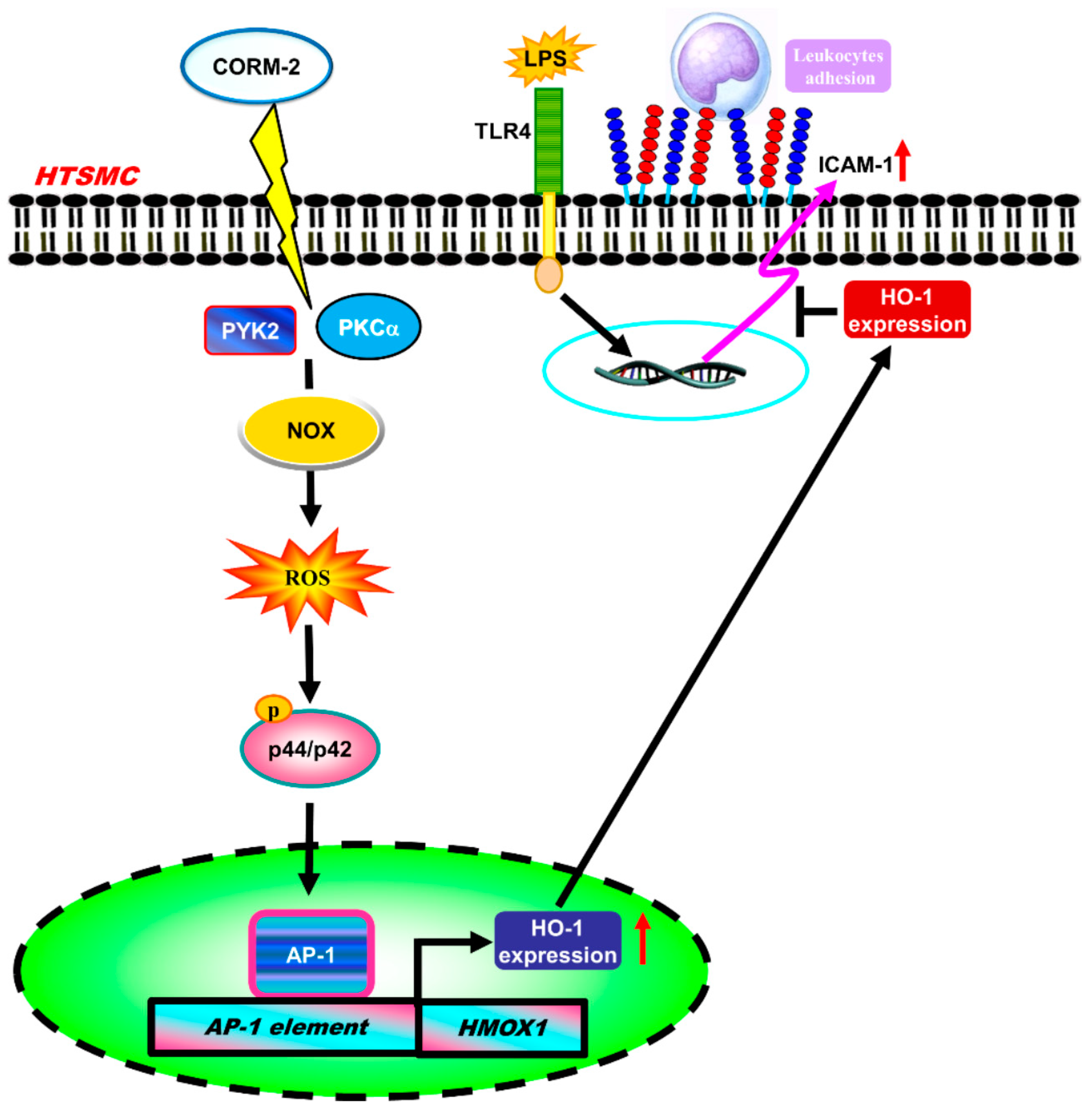
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activator protein 1 |
| APO | apocynin |
| ARE | antioxidant response element |
| BAL | bronchoalveolar lavage |
| BCA | bicinchoninic acid |
| BSA | Bovine serum albumin |
| CO | carbon monoxide |
| CORM-2 | carbon monoxide releasing molecule-2 |
| CoPPIX | cobalt protoporphyrin |
| CSPE | cigarette smoke particle extract |
| DCF | dichlorofluorescein |
| DCF-DA | 2',7'-dichlorofluorescin diacetate |
| DHE | dihydroethidium |
| DMEM/F-12 | Dubecco’s Modified Eagle Medium/Nutrient Mixture F-12 |
| DMSO | dimethyl sulfoxide |
| DPI | diphenyleneiodonium |
| ECL | enhanced chemiluminescence |
| EGFR | epidermal growth factor receptor |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| FBS | fetal bovine serum |
| MCH2DCF-DA | chloromethyl 2’,7’-dichloro fluorescein diacetate |
| H & E | hematoxylin & eosin |
| HO-1 | heme oxygenase-1 |
| HTSMCs | human tracheal smooth muscle cells |
| ICAM-1 | intercellular adhesion molecule |
| ICR | Institute of Cancer Research |
| i.p. | intraperitoneally |
| LPS | lipopolysaccharide |
| LTA | lipotechoic acid |
| MAPKs | mitogen-activated protein kinases |
| mRNA | messenger ribonucleic acid |
| NAC | N-acetyl-cysteine |
| NADPH | nicotinamaide adenine dinucleotide phosphate |
| NF-κB | nuclear factor-κB |
| Nox | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PBS | phosphate-buffered saline |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| Pyk2 | proline-rich tyrosine kinase 2 |
| ROS | reactive oxygen species |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| siRNA | small interfering ribonucleic acid |
| TNF | tumor necrosis factor |
| TSIIA | tanshinone IIA |
| TTBS | Tween-Tris buffered solution |
| VCAM-1 | vascular cell adhesion molecule |
| XTT | 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide |
| ZnPP IX | zinc protoporphyrin IX |
References
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Lee, I.T.; Wang, S.W.; Lee, C.W.; Chang, C.C.; Lin, C.C.; Luo, S.F.; Yang, C.M. Lipoteichoic acid induces HO-1 expression via the TLR2/MyD88/c-Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J. Immunol. 2008, 181, 5098–5110. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [PubMed]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006, 291, 21–28. [Google Scholar] [CrossRef]
- Jamal Uddin, M.; Joe, Y.; Kim, S.K.; Oh Jeong, S.; Ryter, S.W.; Pae, H.O.; Chung, H.T. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell. Mol. Immunol. 2016, 13, 170–179. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Mann, B.E.; Johnson, T.R.; Green, C.J.; Motterlini, R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br. J. Pharmacol. 2005, 145, 8008–8010. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408–4415. [Google Scholar] [CrossRef]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, L.E.; Perrella, M.A.; Mitsialis, S.A. The role of heme oxygenase-1 in pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2007, 36, 158–165. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Devesa, I. Inducers of heme oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Kolls, J.K.; Mantell, L.L.; Cook, J.L.; Alam, J.; Choi, A.M. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J. Clin. Investig. 1999, 103, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Duggal, R.N.; Oliva, O.M.; Karki, S.; Surolia, R.; Wang, Z.; Watson, R.D.; Thannickal, V.J.; Powell, M.; Watts, S.; et al. Heme oxygenase-1 protects corexit 9500A-induced respiratory epithelial injury across species. PLoS ONE 2015, 10, e0122275. [Google Scholar] [CrossRef] [PubMed]
- Srisook, K.; Han, S.S.; Cho, H.S.; Li, M.H.; Ueda, H.; Kim, C.; Cha, Y.N. CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem. Pharmacol. 2006, 71, 307–318. [Google Scholar] [CrossRef]
- Cheng, S.E.; Lee, I.T.; Lin, C.C.; Kou, Y.R.; Yang, C.M. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: Role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic. Biol. Med. 2010, 48, 1410–1422. [Google Scholar] [CrossRef]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell. Longev. 2012, 2012, 794237. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.L.; Lin, C.C.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. CO Induces Nrf2-Dependent Heme Oxygenase-1 Transcription by Cooperating with Sp1 and c-Jun in Rat Brain Astrocytes. Mol. Neurobiol. 2015, 52, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Taille, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Green, C.J.; Foresti, R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid. Redox Signal. 2002, 4, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediat. Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Lin, C.C.; Lee, I.T.; Hsu, C.K.; Tai, Y.C.; Hsieh, H.L.; Chi, P.L.; Hsiao, L.D. c-Src-dependent transactivation of EGFR mediates CORM-2-induced HO-1 expression in human tracheal smooth muscle cells. J. Cell. Physiol. 2015, 230, 2351–2361. [Google Scholar] [CrossRef]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef]
- Constantin, M.; Choi, A.J.; Cloonan, S.M.; Ryter, S.W. Therapeutic potential of heme oxygenase-1/carbon monoxide in lung disease. Int. J. Hypertens. 2012, 2012, 859235. [Google Scholar] [CrossRef]
- Xue, J.; Habtezion, A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J. Clin. Investig. 2014, 124, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhang, J.; Lv, W.; Wang, X.; Sun, B. Effect of carbon monoxide-releasing molecules II-liberated CO on suppressing inflammatory response in sepsis by interfering with nuclear factor kappa B activation. PLoS ONE 2013, 8, e75840. [Google Scholar] [CrossRef] [PubMed]
- Takasuka, H.; Hayashi, S.; Koyama, M.; Yasuda, M.; Aihara, E.; Amagase, K.; Takeuchi, K. Carbon monoxide involved in modulating HCO3− secretion in rat duodenum. J. Pharmacol. Exp. Ther. 2011, 337, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: Refining the toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Lin, C.C.; Chien, P.T.; Hsiao, L.D.; Yan, C.M. Thrombin/Matrix Metalloproteinase-9-Dependent SK-N-SH Cell Migration is Mediated Through a PLC/PKC/MAPKs/NF-kappaB Cascade. Mol. Neurobiol. 2016, 53, 5833–5846. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.D.; Mita, M.; Nakagawa, J.; Shoji, M.; Sutherland, C.; Walsh, M.P. A role for the tyrosine kinase Pyk2 in depolarization-induced contraction of vascular smooth muscle. J. Biol. Chem. 2015, 290, 8677–8692. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Davidson, D.; Souza, C.M.; Vacher, J.; Veillette, A. Macrophage fusion is controlled by the cytoplasmic protein tyrosine phosphatase PTP-PEST/PTPN12. Mol. Cell. Biol. 2013, 33, 2458–2469. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Wang, H.H.; Wu, C.Y.; Yang, C.M. Reactive Oxygen Species-Dependent c-Fos/Activator Protein 1 Induction Upregulates Heme Oxygenase-1 Expression by Bradykinin in Brain Astrocytes. Antioxid. Redox Signal. 2010, 13, 1829–1844. [Google Scholar] [CrossRef]
- Rochette, L.; Cottin, Y.; Zeller, M.; Vergely, C. Carbon monoxide: Mechanisms of action and potential clinical implications. Pharmacol. Ther. 2013, 137, 133–152. [Google Scholar] [CrossRef]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Mulier, B.; Rahman, I.; Watchorn, T.; Donaldson, K.; MacNe, W.; Jeffery, P.K. Hydrogen peroxide-induced epithelial injury: The protective role of intracellular nonprotein thiols (NPSH). Eur. Respir. J. 1998, 11, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, S.S.; Cavalca, V.; Eligini, S.; Brambilla, M.; Caian, A.; Tremoli, E.; Colli, S. Apocynin prevents cyclooxygenase 2 expression in human monocytes through NADPH oxidase and glutathione redox-dependent mechanisms. Free Radic. Biol. Med. 2004, 37, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.; Hynes, A.; Brakebusch, C.; Quondamatteo, F. Rho GTPases and Nox dependent ROS production in skin. Is there a connection? Histol. Histopathol. 2012, 27, 1395–1406. [Google Scholar] [PubMed]
- Tavares, A.F.; Teixeira, M.; Romao, C.C.; Seixas, J.D.; Nobre, L.S.; Saraiva, L.M. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 2011, 286, 26708–26717. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. NO and CO as second messengers in oxygen sensing in the carotid body. Respir. Physiol. 1999, 115, 161–168. [Google Scholar] [CrossRef]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef]
- Alam, R.; Gorska, M.M. Mitogen-activated protein kinase signalling and ERK1/2 bistability in asthma. Clin. Exp. Allergy 2011, 41, 149–159. [Google Scholar] [CrossRef]
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef]
- Foresti, R.; Bani-Hani, M.G.; Motterlini, R. Use of carbon monoxide as a therapeutic agent: Promises and challenges. Intensive Care Med. 2008, 34, 649–658. [Google Scholar] [CrossRef]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Lee, Y.M.; Shih, N.L.; Chen, Y.C.; Yen, M.H. Heme oxygenase-1 contributes to the cytoprotection of alpha-lipoic acid via activation of p44/42 mitogen-activated protein kinase in vascular smooth muscle cells. Free Radic. Biol. Med. 2006, 40, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933. [Google Scholar] [PubMed]
- Urquhart, P.; Rosignoli, G.; Cooper, D.; Motterlini, R.; Perretti, M. Carbon monoxide-releasing molecules modulate leukocyte-endothelial interactions under flow. J. Pharmacol. Exp. Ther. 2007, 321, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Almolki, A.; Taillé, C.; Martin, G.F.; Jose, P.J.; Zedda, C.; Conti, M.; Megret, J.; Henin, D.; Aubier, M.; Boczkowski, J. Heme oxygenase attenuates allergen-induced airway inflammation and hyperreactivity in guinea pigs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L26–L34. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Sun, H.; Liu, C.; Shen, J.; Chen, Z.; Chen, X. Role of CO-releasing molecules liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. J. Surg. Res. 2007, 139, 128–135. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; Group NCRRGW. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.C.; Li, X.H.; McRobb, L.S.; Heather, A.K. Inhibitory Effect of a French Maritime Pine Bark Extract-Based Nutritional Supplement on TNF-alpha-Induced Inflammation and Oxidative Stress in Human Coronary Artery Endothelial Cells. Evid. Based Complement. Altern. Med. 2015, 2015, 260530. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Lee, I.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Nox2/ROS-dependent human antigen R translocation contributes to TNF-alpha-induced SOCS-3 expression in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L521–L533. [Google Scholar] [CrossRef]
- Lee, C.W.; Chien, C.S.; Yang, C.M. Lipoteichoic acid-stimulated p42/p44 MAPK activation via Toll-like receptor 2 in tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L921–L930. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. High glucose induces reactive oxygen species-dependent matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Mol. Neurobiol. 2013, 48, 601–614. [Google Scholar] [CrossRef] [PubMed]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-C.; Hsiao, L.-D.; Cho, R.-L.; Yang, C.-M. Carbon Monoxide Releasing Molecule-2-Upregulated ROS-Dependent Heme Oxygenase-1 Axis Suppresses Lipopolysaccharide-Induced Airway Inflammation. Int. J. Mol. Sci. 2019, 20, 3157. https://doi.org/10.3390/ijms20133157
Lin C-C, Hsiao L-D, Cho R-L, Yang C-M. Carbon Monoxide Releasing Molecule-2-Upregulated ROS-Dependent Heme Oxygenase-1 Axis Suppresses Lipopolysaccharide-Induced Airway Inflammation. International Journal of Molecular Sciences. 2019; 20(13):3157. https://doi.org/10.3390/ijms20133157
Chicago/Turabian StyleLin, Chih-Chung, Li-Der Hsiao, Rou-Ling Cho, and Chuen-Mao Yang. 2019. "Carbon Monoxide Releasing Molecule-2-Upregulated ROS-Dependent Heme Oxygenase-1 Axis Suppresses Lipopolysaccharide-Induced Airway Inflammation" International Journal of Molecular Sciences 20, no. 13: 3157. https://doi.org/10.3390/ijms20133157
APA StyleLin, C.-C., Hsiao, L.-D., Cho, R.-L., & Yang, C.-M. (2019). Carbon Monoxide Releasing Molecule-2-Upregulated ROS-Dependent Heme Oxygenase-1 Axis Suppresses Lipopolysaccharide-Induced Airway Inflammation. International Journal of Molecular Sciences, 20(13), 3157. https://doi.org/10.3390/ijms20133157