Virtual Screening Guided Design, Synthesis and Bioactivity Study of Benzisoselenazolones (BISAs) on Inhibition of c-Met and Its Downstream Signalling Pathways


 ,
,
Abstract
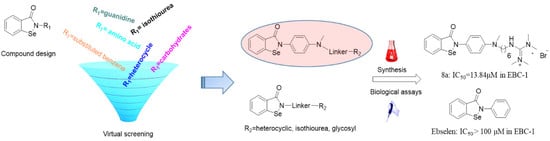
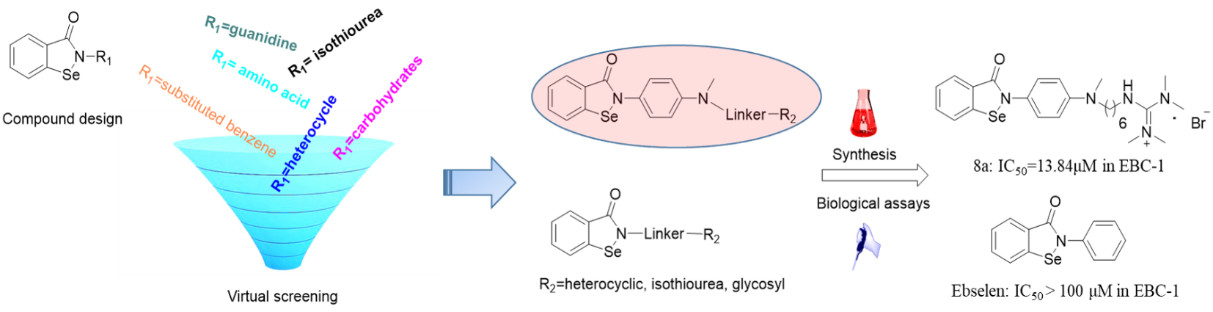
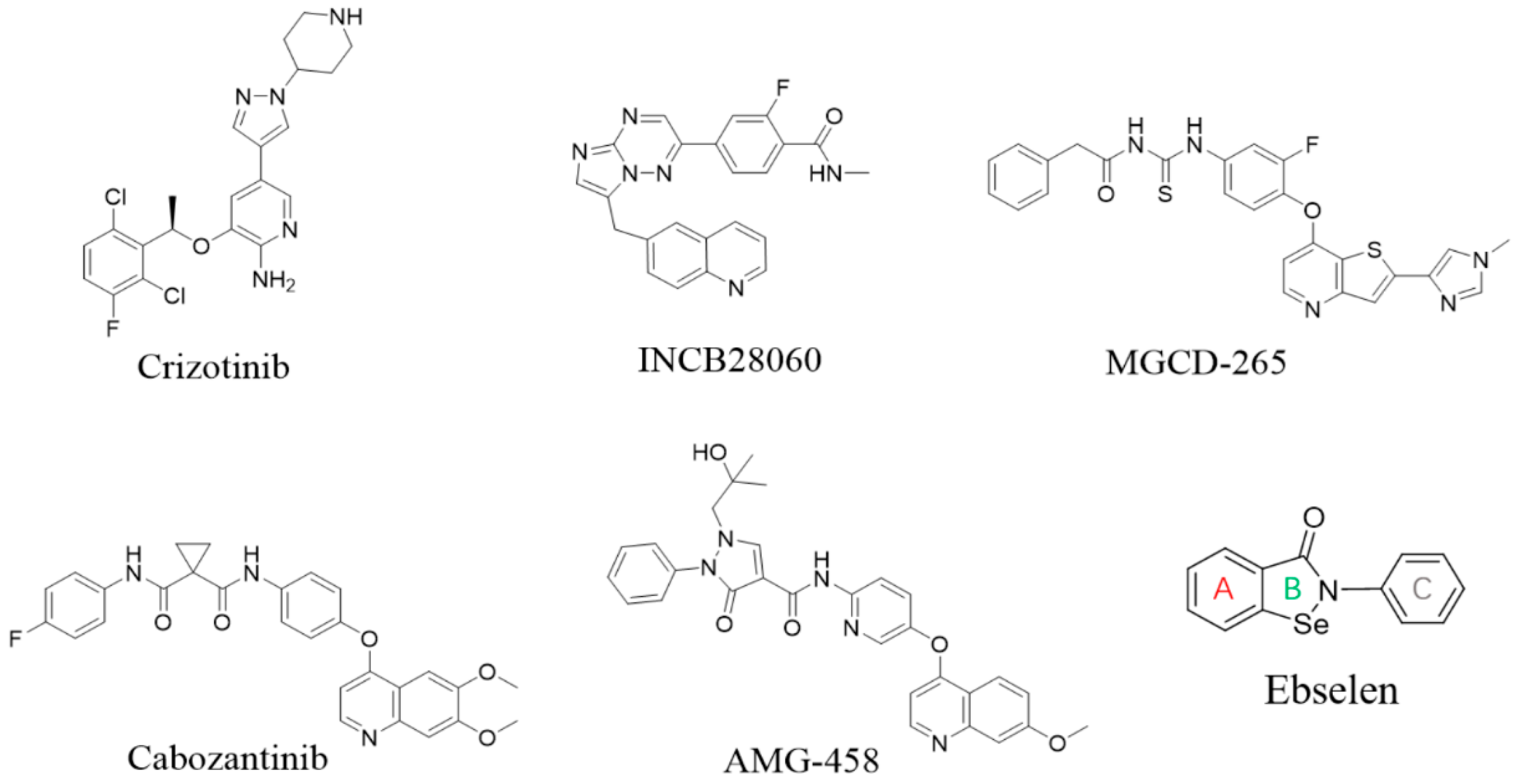
1. Introduction
2. Results
2.1. Virtual Screening and Binding Energy Calculations
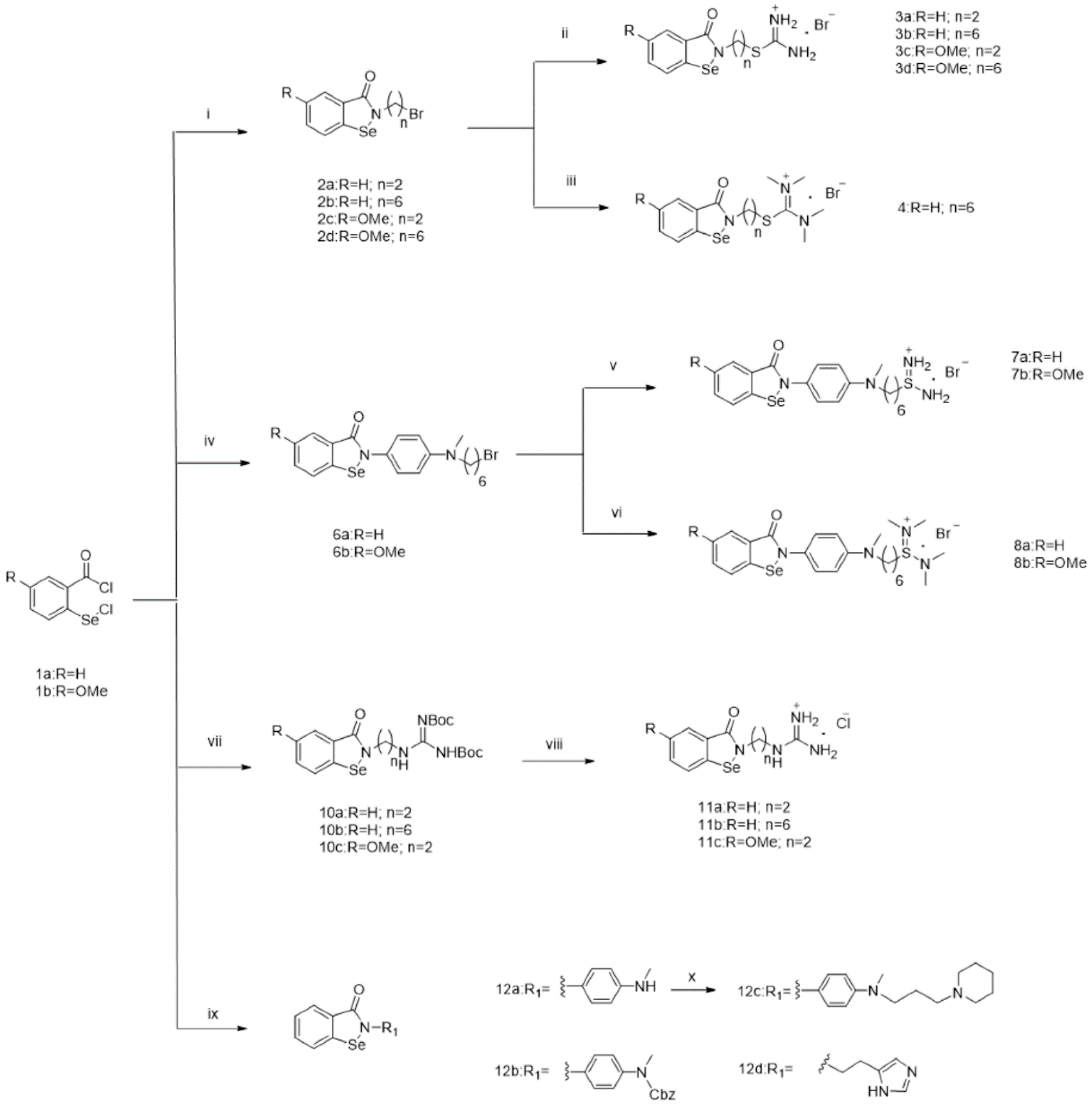
2.2. Chemistry
2.3. Biology Assays
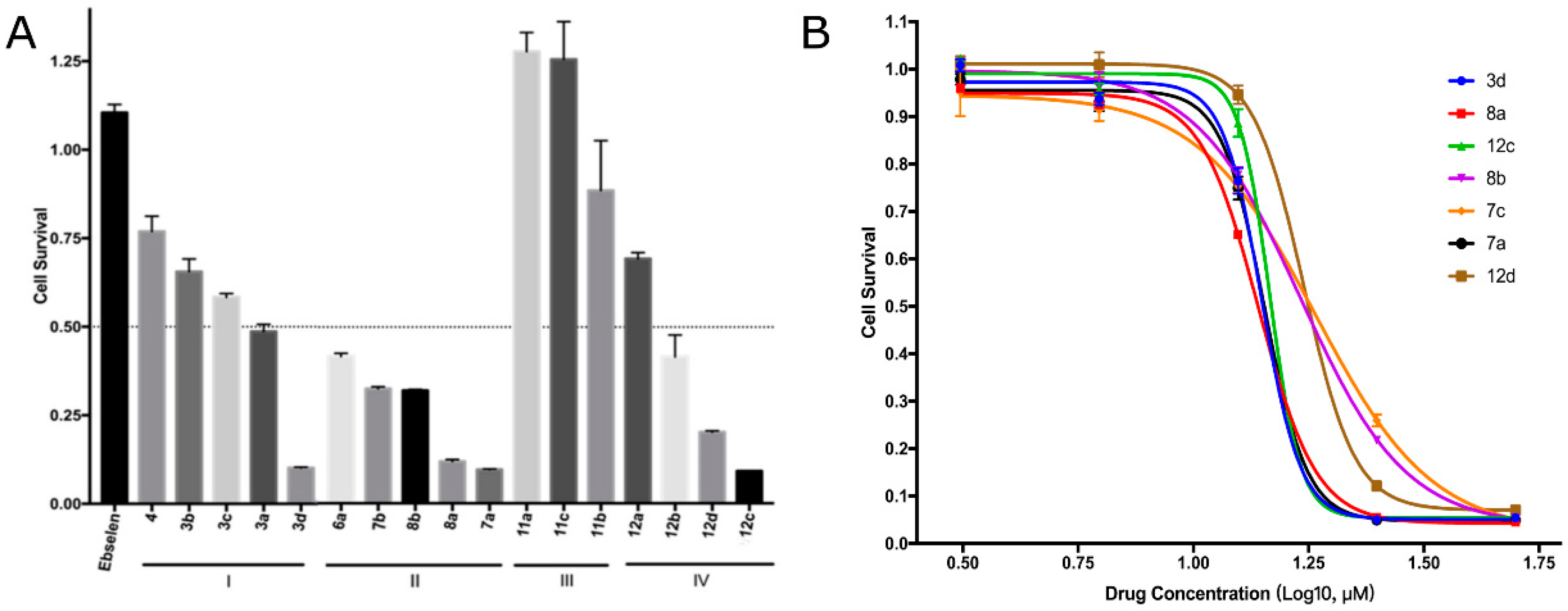
2.3.1. EBC-1 Cell Viability Inhibition
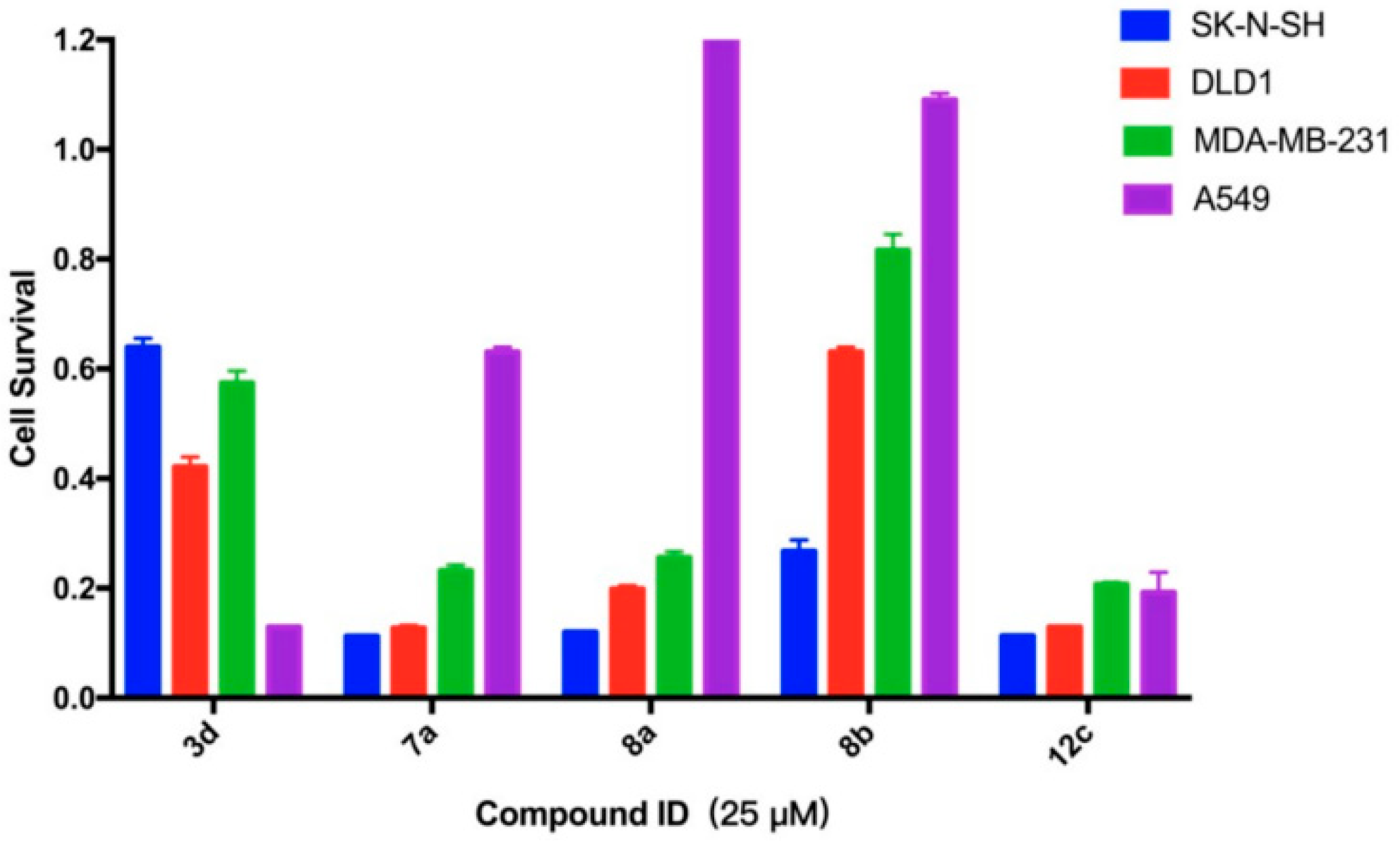
2.3.2. Survival Inhibition of Four Other Cancer Cell Lines
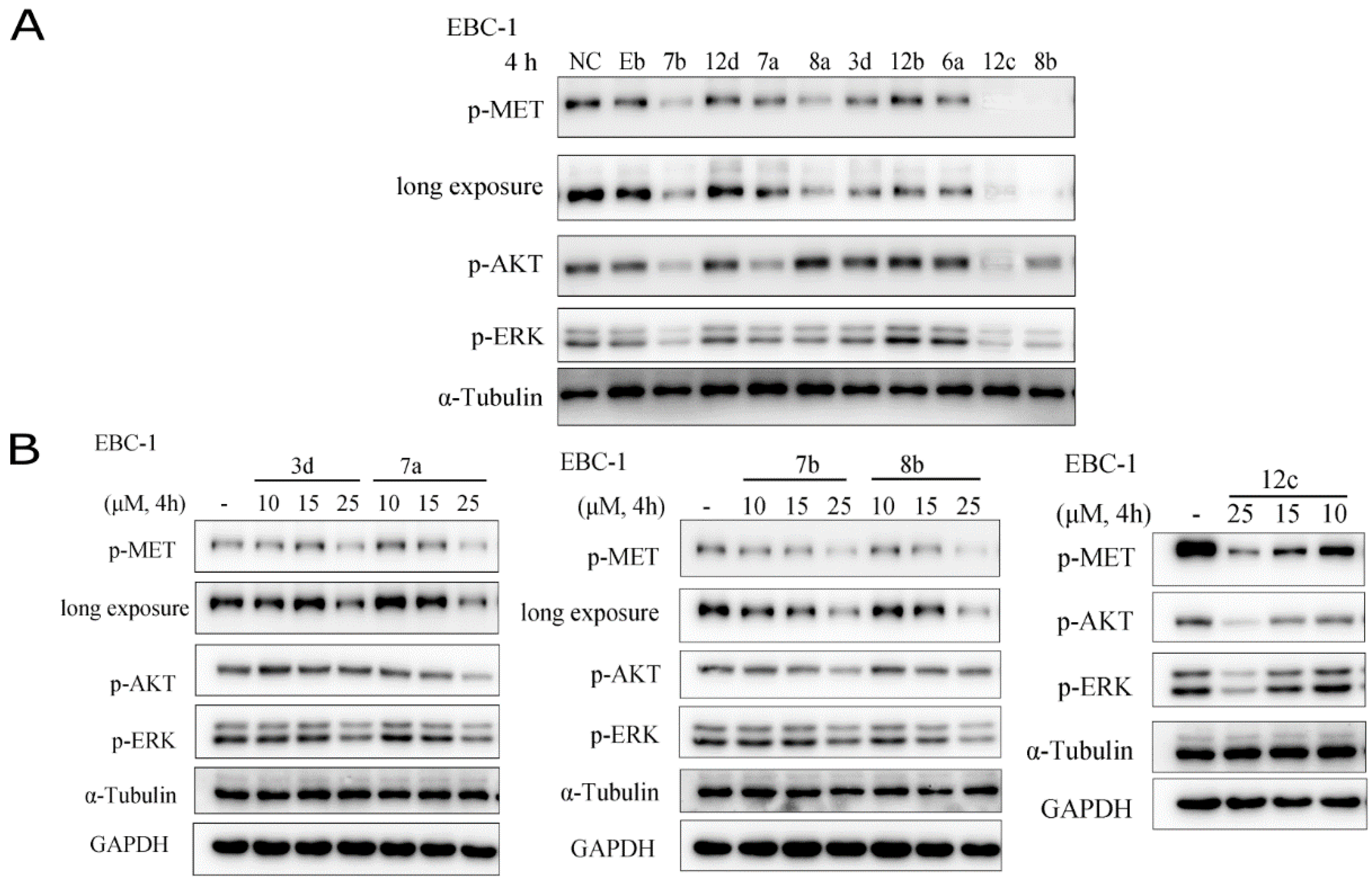
2.3.3. The inhibitory effects on the activation of c-Met and its downstream signalling pathways
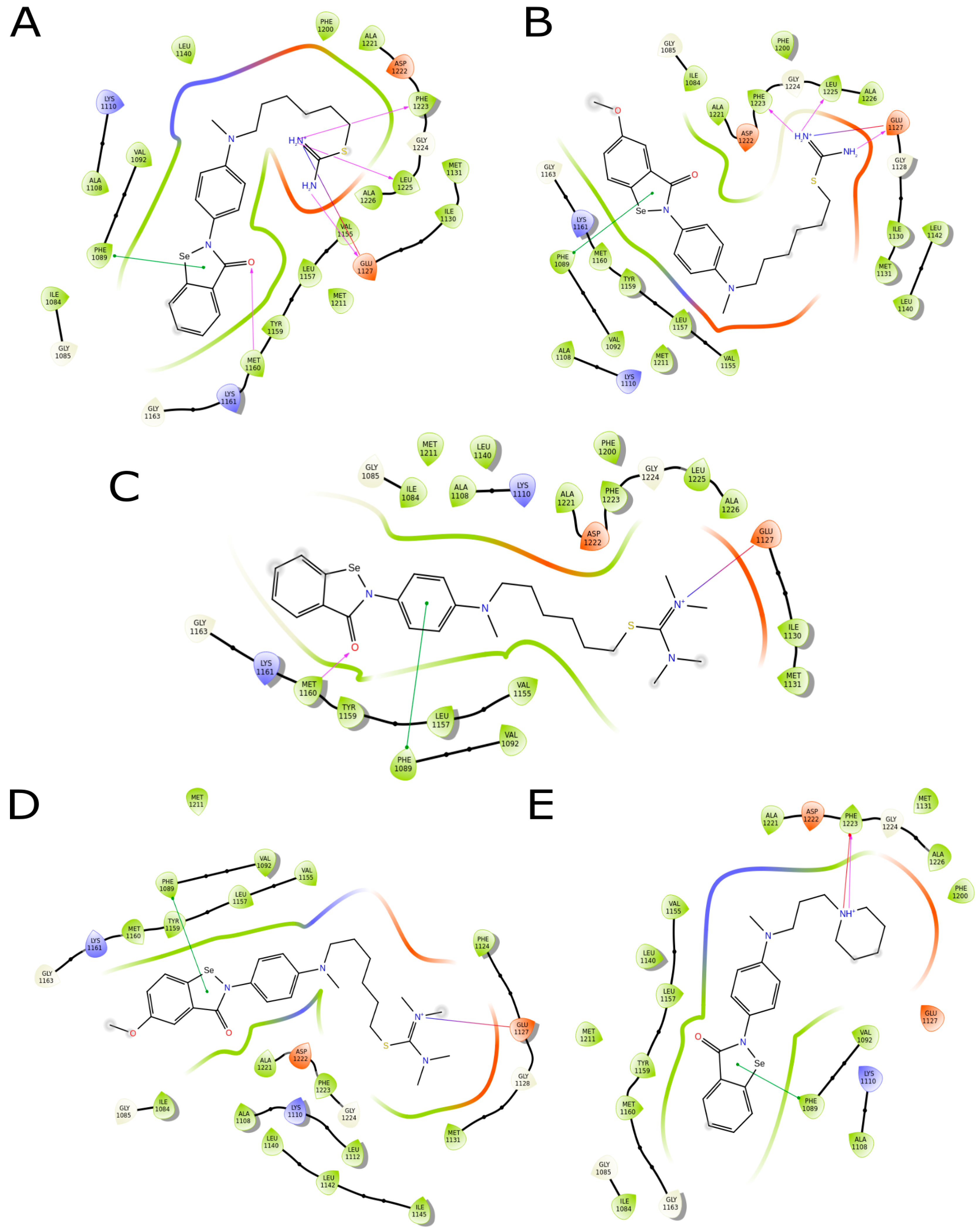
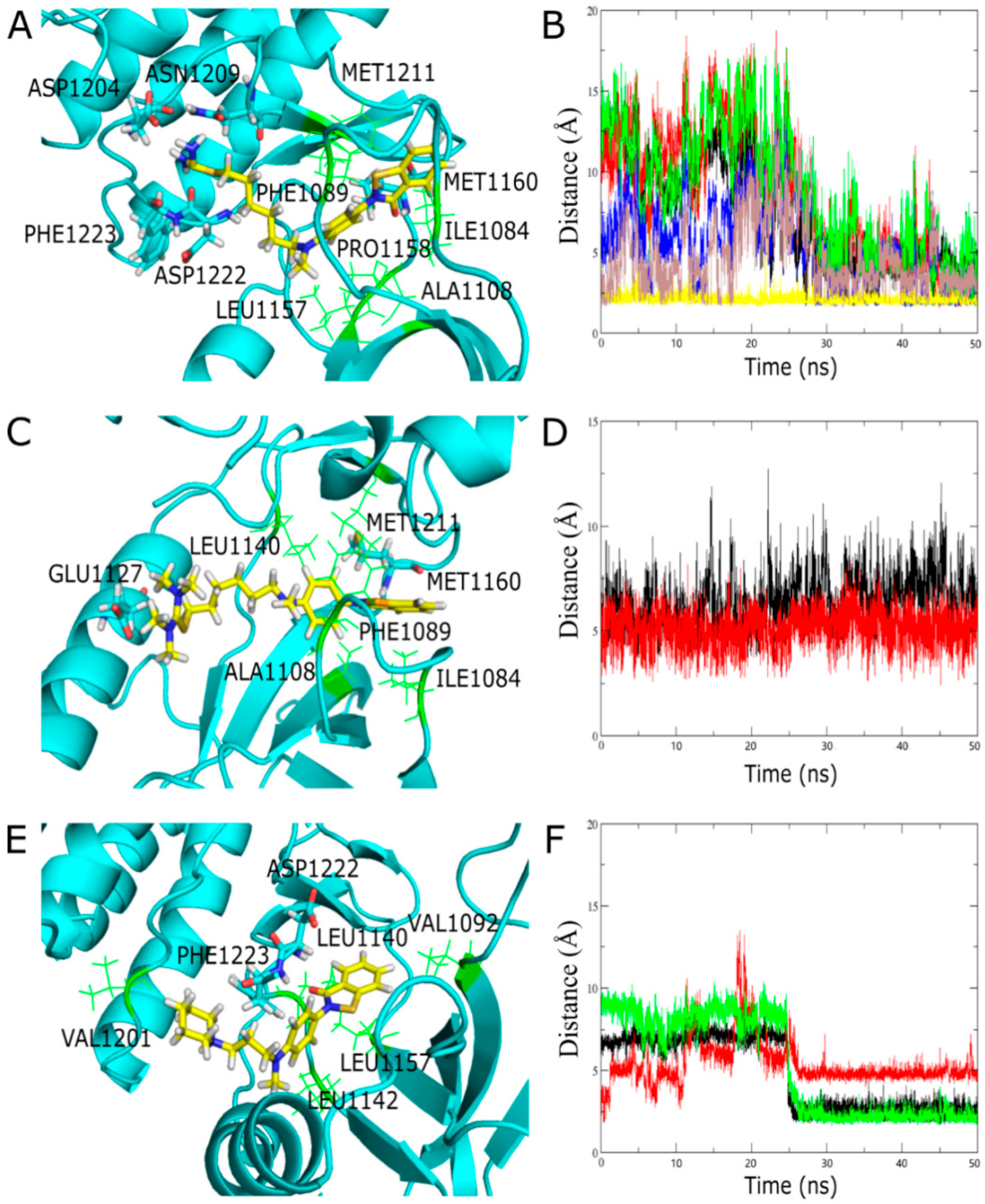
2.3.4. Molecule Docking Studies and Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Virtual Screening and Binding Energy Calculations
4.2. Chemical Synthesis
4.2.1. General Procedure for Compounds 2a~d
4.2.2. General Procedure for Compounds 3a~d
4.2.3. Synthesis of Compound (4)
4.2.4. Synthesis of Compound (5)
4.2.5. General Procedure for Compounds 6a~b
4.2.6. General Procedure for Compounds 7a~b
4.2.7. General Procedure for Compounds 8a~b
4.2.8. General Procedure for Compounds 9a~b
4.2.9. General Procedure for Compounds 10a~c
4.2.10. General Procedure for Compounds 11a~c
4.2.11. Synthesis of Compound (12a)
4.2.12. Synthesis of Compound (12b)
4.2.13. Synthesis of Compound (12c)
4.2.14. Synthesis of Compound (12d)
4.3. Resazurin Assay
4.4. Western Blot
4.5. Molecular Docking
4.6. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGF | Hepatocyte growth factor |
| c-Met | Cellular mesenchymal–epithelial transition factor |
| RON | Recepteur d’Origine Nantais |
| TK | Tyrosine kinase |
| TKIs | Tyrosine kinase inhibitors |
| BISAs | Benzisoselenazolones |
| FAK | Focal Adhesion Kinase |
| AKT | Protein kinase B |
| ERK | Extracellular regulated protein kinases |
| ATP | Adenosine triphosphate |
| THF | Tetrahydrofuran |
| DCM | Dichloromethane |
| DMSO | Dimethyl sulphoxide |
| RMSD | Root mean square deviation |
References
- Fajardo-Puerta, A.B.; Mato Prado, M.; Frampton, A.E.; Jiao, L.R. Gene of the month: HGF. J. Clin. Pathol. 2016, 69, 575–579. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, A.; Ryu, J.Y.; Cho, Y.J.; Choi, J.J.; Song, S.Y.; Bang, H.; Lee, J.S.; Cho, W.C.; Choi, C.H.; et al. c-MET as a Potential Therapeutic Target in Ovarian Clear Cell Carcinoma. Sci. Rep. 2016, 6, 38502. [Google Scholar] [CrossRef]
- Yang, Y.F.; Zhang, Y.; Yang, L.Y.; Zhao, L.L.; Si, L.H.; Zhang, H.B.; Liu, Q.S. Discovery of imidazopyridine derivatives as novel c-Met kinase inhibitors: Synthesis, SAR study, and biological activity. Bioorg. Chem. 2017, 70, 126–132. [Google Scholar] [CrossRef]
- Chen, G.Z.; Dai, W.S.; Zhu, H.C.; Song, H.M.; Yang, X.; Wang, Y.D.; Min, H.; Lu, Q.; Liu, S.; Sun, X.C.; et al. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J. Cancer 2017, 8, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An Orally Available Small-Molecule Inhibitor of c-Met, PF-2341066, Exhibits Cytoreductive Antitumor Efficacy through Antiproliferative and Antiangiogenic Mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef]
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A.; et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005, 65, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Van Leenders, G.J.; Sookhlall, R.; Teubel, W.J.; de Ridder, C.M.; Reneman, S.; Sacchetti, A.; Vissers, K.J.; van Weerden, W.; Jenster, G. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS ONE 2011, 6, e26753. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, M.N.; Jeong, B.C.; Oh, S.H.; Kook, M.S.; Koh, J.T. Chemical inhibitors of c-Met receptor tyrosine kinase stimulate osteoblast differentiation and bone regeneration. Eur. J. Pharmacol. 2017, 806, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Varkaris, A.; Corn, P.G.; Gaur, S.; Dayyani, F.; Logothetis, C.J.; Gallick, G.E. The role of HGF/c-Met signaling in prostate cancer progression and c-Met inhibitors in clinical trials. Expert. Opin. Investig. Drugs 2011, 20, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Wu, G.Z.; Kaas, Q.; Jiang, T.; Yu, R.L. Development of Efficient Docking Strategies and Structure-activity Relationship Study of the c-Met Type II Inhibitors. J. Mol. Graph. Model. 2017, 75, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Jalbout, A.F.; Hameed, A.J.; Essa, A.H. Structural isomers of 2-(2,3 and 4-substituted-phenyl)-1,2-benzisoselenazol-3(2H)-one: A Theoretical Study. J. Organomet. Chem. 2008, 693, 2074–2078. [Google Scholar] [CrossRef]
- Luo, Z.; Sheng, J.; Sun, Y.; Lu, C.; Yan, J.; Liu, A.; Luo, H.B.; Huang, L.; Li, X. Synthesis and Evaluation of Multi-Target-Directed Ligands against Alzheimer’s Disease Based on the Fusion of Donepezil and Ebselen. J. Med. Chem. 2013, 56, 9089–9099. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Biedermann, J.; Bittner, C.; Dereu, N.; Leyck, S.; Wetzig, H. Structure-activity relationships of a series of anti-inflammatory benzisoselenazolones (BISAs). Agents Act. 1989, 27, 306–308. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, Z.; Chen, N.; Shi, Q.; Tong, L.; Kong, F.; Cheng, X.; Chen, H.; Wang, C.; Tang, B. Target Discovery of Ebselen with a Biotinylated Probe. Chem. Commun. (Camb.) 2018, 54, 9506–9509. [Google Scholar] [CrossRef]
- Wendel, A.; Fausel, M.; Safayhi, H.; Tiegs, G.; Otter, R. A novel biologically active seleno-organic compound--II. Activity of PZ 51 in relation to glutathione peroxidase. Biochem. Pharmacol. 1984, 33, 3241–3245. [Google Scholar] [CrossRef]
- Maiorino, M.; Roveri, A.; Coassin, M.; Ursini, F. Kinetic mechanism and substrate specificity of glutathione peroxidase activity of ebselen (PZ51). Biochem. Pharmacol. 1988, 37, 2267–2271. [Google Scholar] [CrossRef]
- Masumoto, H.; Kissner, R.; Koppenol, W.H.; Sies, H. Kinetic study of the reaction of ebselen with peroxynitrite. FEBS Lett. 1996, 398, 179–182. [Google Scholar] [CrossRef]
- Terentis, A.C.; Freewan, M.; Sempertegui Plaza, T.S.; Raftery, M.J.; Stocker, R.; Thomas, S.R. The selenazal drug ebselen potently inhibits indoleamine 2,3-dioxygenase by targeting enzyme cysteine residues. Biochemistry 2010, 49, 591–600. [Google Scholar] [CrossRef]
- Mugesh, G.; du Mont, W.W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef]
- Bijian, K.; Zhang, Z.; Xu, B.; Jie, S.; Chen, B.; Wan, S.; Wu, J.; Jiang, T.; Alaoui-Jamali, M.A. Synthesis and biological activity of novel organoselenium derivatives targeting multiple kinases and capable of inhibiting cancer progression to metastases. Eur. J. Med. Chem. 2012, 48, 143–152. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, S.; Chang, Y.; Fan, D.; Agostini, A.; Zhang, L.; Jiang, T. The Aglycone Ebselen and β-D-Xyloside Primed Glycosaminoglycans Co-contribute to Ebselen β-D-Xyloside-induced Cytotoxicity. J. Med. Chem. 2018, 61, 2937–2948. [Google Scholar] [CrossRef]
- Bean, G.J.; Flickinger, S.T.; Westler, W.M.; McCully, M.E.; Sept, D.; Weibel, D.B.; Amann, K.J. A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry 2009, 48, 4852–4857. [Google Scholar] [CrossRef]
- Alcolea, V.; Plano, D.; Karelia, D.N.; Palop, J.A.; Amin, S.; Sanmartín, C.; Sharma, A.K. Novel seleno- and thio-urea derivatives with potent in vitro activities against several cancer cell lines. Eur. J. Med. Chem. 2016, 113, 134–144. [Google Scholar] [CrossRef]
- Wang, H.; Yan, C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia 2011, 13, 611–619. [Google Scholar] [CrossRef]
- Tong, S.; Zhang, M.; Wang, S.; Yin, R.; Yu, R.; Wan, S.; Jiang, T.; Zhang, L. Isothiouronium modification empowers pyrimidine-substituted curcumin analogs potent cytotoxicity and Golgi localization. Eur. J. Med. Chem. 2016, 123, 849–857. [Google Scholar] [CrossRef]
- Cui, J.J.; McTigue, M.; Nambu, M.; Tran-Dubé, M.; Pairish, M.; Shen, H.; Jia, L.; Cheng, H.; Hoffman, J.; Le, P.; et al. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 2012, 55, 8091–8109. [Google Scholar]
- Norman, M.H.; Liu, L.; Lee, M.; Xi, N.; Fellows, I.; D’Angelo, N.D.; Dominguez, C.; Rex, K.; Bellon, S.F.; Kim, T.S.; et al. Structure-based design of novel class II c-Met inhibitors: 1. Identification of pyrazolone-based derivatives. J. Med. Chem. 2012, 55, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Verkhivker, G.M. Integrating ligand-based and protein-centric virtual screening of kinase inhibitors using ensembles of multiple protein kinase genes and conformations. J. Chem. Inf. Model. 2012, 52, 2501–2515. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 2015, 20, 720–729. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Jiang, J.; Murphy, M.P.; Epperly, M.; Zhang, X.; Li, S.; Greenberger, J.; Kagan, V.; Bayır, H. Design and Synthesis of a Mitochondria-Targeted Mimic of Glutathione Peroxidase, MitoEbselen-2, as a Radiation Mitigator. ACS Med. Chem. Lett. 2014, 5, 1304. [Google Scholar] [CrossRef]
- Bénard, S.; Neuville, L.; Zhu, J. Copper-promoted N-cyclopropylation of anilines and amines by cyclopropylboronic acid. Chem. Commun. (Camb.) 2010, 46, 3393–3395. [Google Scholar]
- Egger, J.; Weckerle, C.; Cutting, B.; Schwardt, O.; Rabbani, S.; Lemme, K.; Ernst, B. Nanomolar E-selectin antagonists with prolonged half-lives by a fragment-based approach. J. Am. Chem. Soc. 2013, 135, 9820–9828. [Google Scholar] [CrossRef]
- Awuah, E.; Ma, E.; Hoegl, A.; Vong, K.; Habib, E.; Auclair, K. Exploring structural motifs necessary for substrate binding in the active site of Escherichia coli pantothenate kinase. Bioorg. Med. Chem. 2014, 22, 3083–3090. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Sun, X.; Song, Q.; He, L.; Yan, L.; Liu, J.; Zhang, Q.; Yu, Q. Receptor Tyrosine Kinase Phosphorylation Pattern-Based Multidrug Combination Is an Effective Approach for Personalized Cancer Treatment. Mol. Cancer Ther. 2016, 15, 2508–2520. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Sköldenberg, E.G.; Larsson, A.; Jakobson, A.; Hedborg, F.; Kogner, P.; Christofferson, R.H.; Azarbayjani, F. The angiogenic growth factors HGF and VEGF in serum and plasma from neuroblastoma patients. Anticance. Res. 2009, 29, 3311–3319. [Google Scholar]
- Kim, D.; Kim, S.; Koh, H.; Yoon, S.O.; Chung, A.S.; Cho, K.S.; Chung, J. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001, 15, 1953. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug. Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Hickey, S.M.; Ashton, T.D.; Pfeffer, F.M. Facile Synthesis of Guanidine Functionalised Building Blocks. Asian J. Org. Chem. 2015, 4, 320–326. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | n | R2 | IC50 of Cell Viability (μM) |
|---|---|---|---|---|
| 3a | H | 2 |  | >25 |
| 3b | H | 6 |  | >25 |
| 3c | OMe | 2 |  | >25 |
| 3d | OMe | 6 |  | 14.09 |
| 4 | H | 6 |  | >25 |
| 6a | H | 6 | Br | >25 |
| 7a | H | 6 |  | 14.31 |
| 7b | OMe | 6 |  | 17.29 |
| 8a | H | 6 |  | 13.84 |
| 8b | OMe | 6 |  | 16.79 |
| 11a | H | 2 |  | >50 |
| 11b | H | 6 |  | >50 |
| 11c | OMe | 2 |  | >50 |
| 12a | H | / |  | >25 |
| 12b | H | / |  | >25 |
| 12c | H | / |  | 14.82 |
| 12d | H | / |  | 17.28 |
| Compound | IC50 values of cell viability (μM) | ||||
|---|---|---|---|---|---|
| SK-N-SH | DLD1 | MDA-MB-231 | A549 | EBC-1 | |
| 3d | 26.55 | 20.61 | 23.18 | 14.91 | 14.09 |
| 7a | 14.83 | 14.95 | 15.50 | 28.84 | 14.31 |
| 8a | 11.51 | 16.48 | 16.41 | 45.68 | 13.84 |
| 8b | 17.32 | 28.84 | 31.75 | 101.6 | 16.79 |
| 12c | 13.86 | 14.91 | 22.63 | 23.19 | 14.82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Song, Q.; Wang, X.; Wei, Z.; Yu, R.; Wang, X.; Jiang, T. Virtual Screening Guided Design, Synthesis and Bioactivity Study of Benzisoselenazolones (BISAs) on Inhibition of c-Met and Its Downstream Signalling Pathways. Int. J. Mol. Sci. 2019, 20, 2489. https://doi.org/10.3390/ijms20102489
Zhang S, Song Q, Wang X, Wei Z, Yu R, Wang X, Jiang T. Virtual Screening Guided Design, Synthesis and Bioactivity Study of Benzisoselenazolones (BISAs) on Inhibition of c-Met and Its Downstream Signalling Pathways. International Journal of Molecular Sciences. 2019; 20(10):2489. https://doi.org/10.3390/ijms20102489
Chicago/Turabian StyleZhang, Siqi, Qiaoling Song, Xueting Wang, Zhiqiang Wei, Rilei Yu, Xin Wang, and Tao Jiang. 2019. "Virtual Screening Guided Design, Synthesis and Bioactivity Study of Benzisoselenazolones (BISAs) on Inhibition of c-Met and Its Downstream Signalling Pathways" International Journal of Molecular Sciences 20, no. 10: 2489. https://doi.org/10.3390/ijms20102489
APA StyleZhang, S., Song, Q., Wang, X., Wei, Z., Yu, R., Wang, X., & Jiang, T. (2019). Virtual Screening Guided Design, Synthesis and Bioactivity Study of Benzisoselenazolones (BISAs) on Inhibition of c-Met and Its Downstream Signalling Pathways. International Journal of Molecular Sciences, 20(10), 2489. https://doi.org/10.3390/ijms20102489