Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It



Abstract
1. Introduction
2. Importance of the cHL Tumor Microenvironment
2.1. TME Cellular Composition
2.2. TME Formation
2.3. TME Composition as a Prognostic Factor
3. HRS Cell-Mediated Immune Escape
4. TME-Mediated Immune Escape
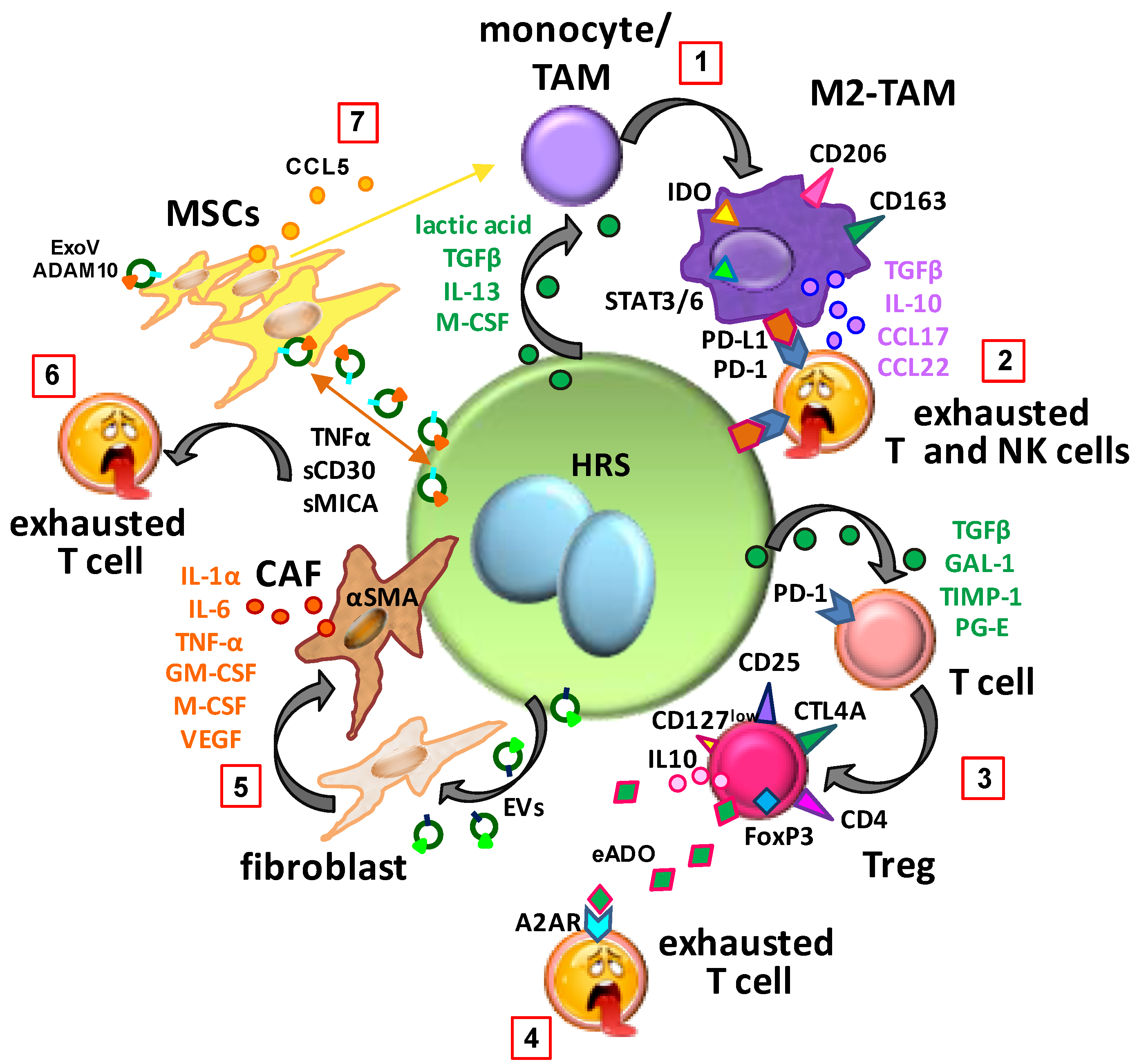
5. Immunosuppressive Education of Normal Cells in the TME
5.1. Monocyte Polarization Towards M2-TAM
5.2. T Cell Polarization towards Immunosuppressive Tregs
5.3. Education of MSCs
5.4. Education by Extracellular Vesicles
6. Targeting the TME to Counteract Its Tumor-Protective Effects
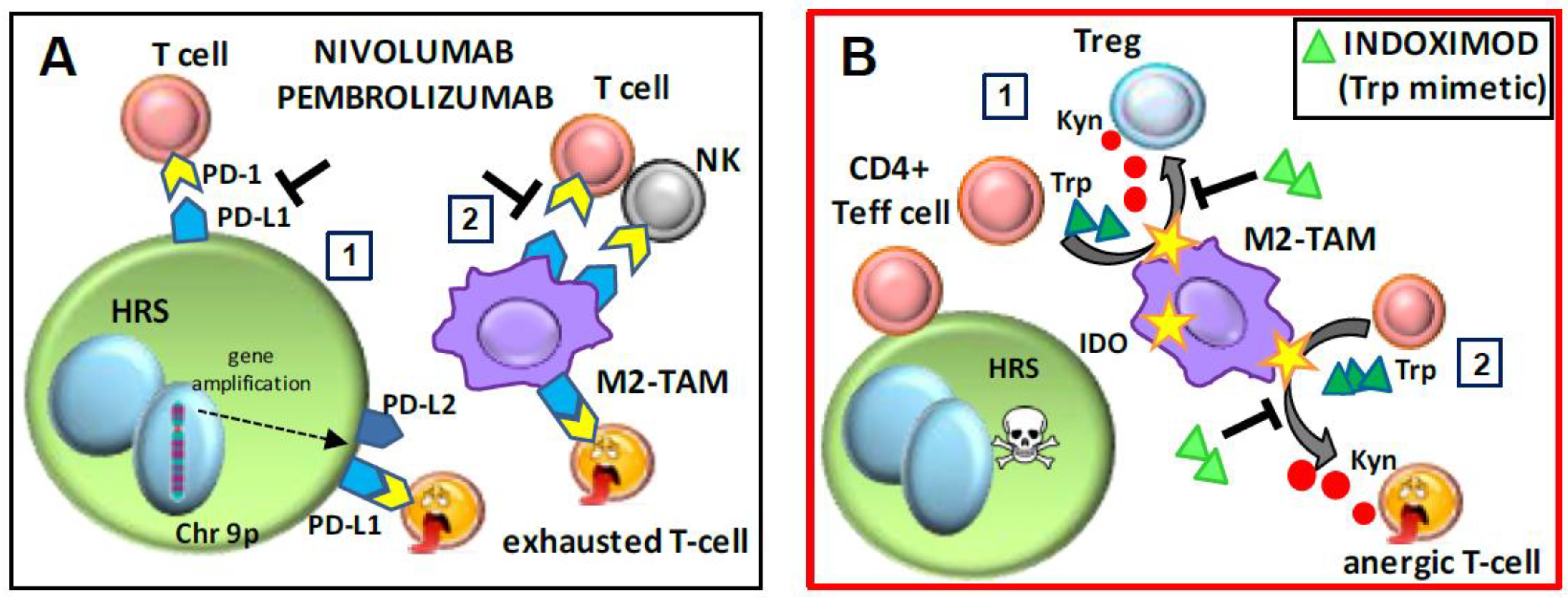
6.1. Checkpoint Inhibitors and Adjuvants: Nivolumab, Pembrolizumab, and Indoximod
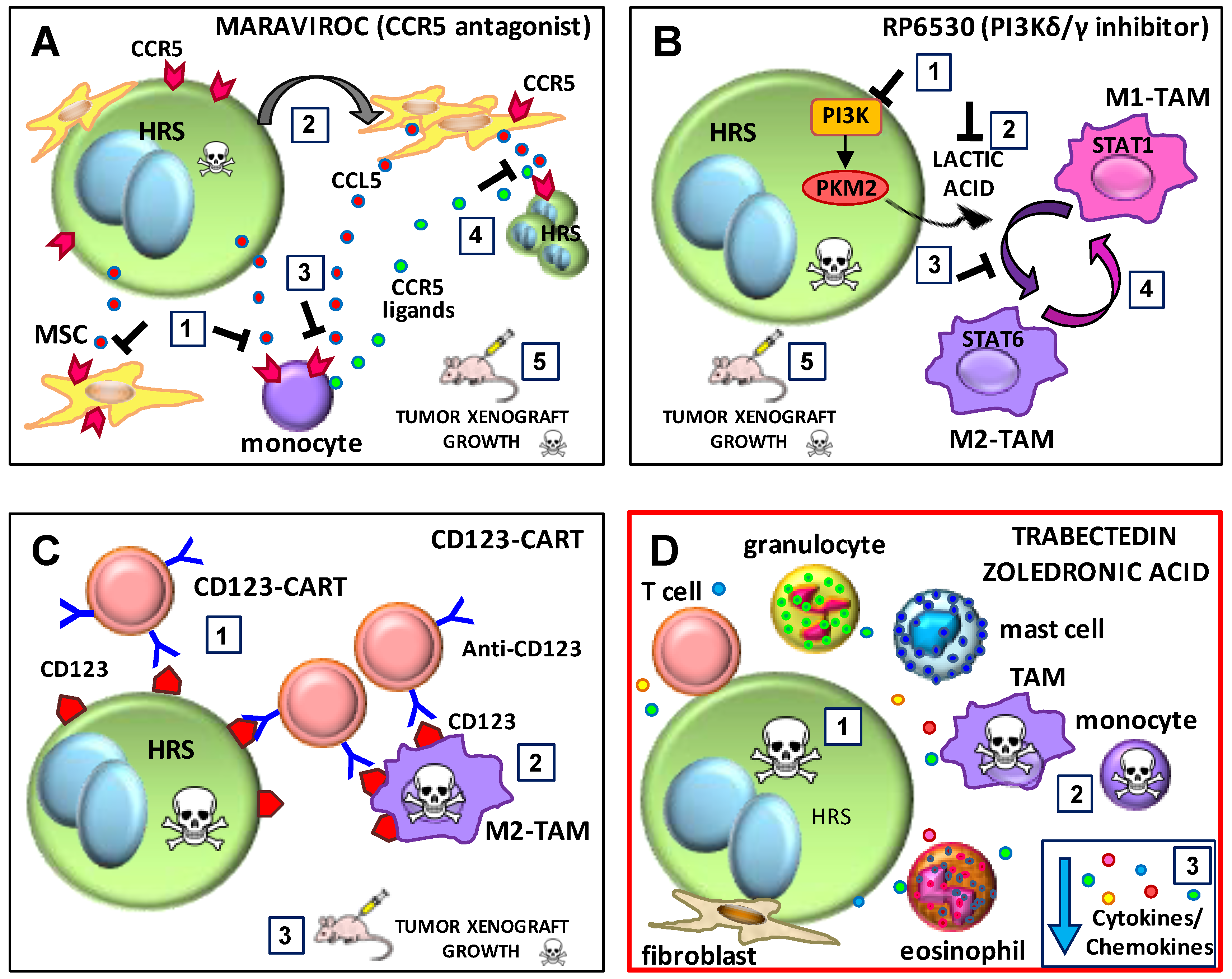
6.2. The CCR5 Antagonist Maraviroc
6.3. The PI3K-δ/ϒ Inhibitor RP6530
6.4. CD123-CAR T Cells
6.5. Trabectedin and Zoledronic Acid
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ansell, S.M. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Mottok, A.; Steidl, C. Biology of classical Hodgkin lymphoma: Implications for prognosis and novel therapies. Blood 2018, 131, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Xavier de, C.A.; Maiato, H.; Maia, A.F.; Ribeiro, S.A.; Pontes, P.; Bickmore, W.; Earnshaw, W.C.; Sambade, C. Reed-sternberg cells form by abscission failure in the presence of functional aurora B kinase. PLoS One. 2015, 10, e0124629. [Google Scholar]
- Rengstl, B.; Newrzela, S.; Heinrich, T.; Weiser, C.; Thalheimer, F.B.; Schmid, F.; Warner, K.; Hartmann, S.; Schroeder, T.; Kuppers, R.; et al. Incomplete cytokinesis and re-fusion of small mononucleated Hodgkin cells lead to giant multinucleated Reed-Sternberg cells. Proc. Natl. Acad. Sci. U S A. 2013, 110, 20729–20734. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.R.; Shipp, M.A. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 2017, 130, 2265–2270. [Google Scholar]
- Hollander, P.; Rostgaard, K.; Smedby, K.E.; Molin, D.; Loskog, A.; de Nully, B.P.; Enblad, G.; Amini, R.M.; Hjalgrim, H.; Glimelius, I. An anergic immune signature in the tumor microenvironment of classical Hodgkin lymphoma is associated with inferior outcome. Eur. J. Haematol. 2018, 100, 88–97. [Google Scholar]
- Tiacci, E.; Doring, C.; Brune, V.; van Noesel, C.J.; Klapper, W.; Mechtersheimer, G.; Falini, B.; Kuppers, R.; Hansmann, M.L. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 2012, 120, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Engert, A.; Hansmann, M.L. Hodgkin lymphoma. J. Clin. Invest. 2012, 122, 3439–3447. [Google Scholar] [PubMed]
- Kreher, S.; Bouhlel, M.A.; Cauchy, P.; Lamprecht, B.; Li, S.; Grau, M.; Hummel, F.; Kochert, K.; Anagnostopoulos, I.; Johrens, K.; et al. Mapping of transcription factor motifs in active chromatin identifies IRF5 as key regulator in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. U S A. 2014, 111, E4513–E4522. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Aldinucci, D.; Gattei, V.; Dalla-Favera, R.; Gaidano, G. Expression pattern of MUM1/IRF4 in the spectrum of pathology of Hodgkin’s disease. Br. J. Haematol. 2002, 117, 366–372. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. Hodgkin lymphoma classification: Are we at a crossroads? Cancer 2017, 123, 3654–3655. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.C.; McAulay, K.A.; Montgomery, D.; Lake, A.; Shield, L.; Gallagher, A.; Little, A.M.; Shah, A.; Marsh, S.G.; Taylor, G.M.; et al. Modeling HLA associations with EBV-positive and -negative Hodgkin lymphoma suggests distinct mechanisms in disease pathogenesis. Int. J. Cancer. 2015, 137, 1066–1075. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A. Epstein Barr Virus-Associated Hodgkin Lymphoma. Cancers (Basel) 2018, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Carbone, A.; Dolcetti, R. Microenvironment and HIV-related lymphomagenesis. Semin Cancer Biol. 2015, 34, 52–57. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; Li, X.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer. 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, F.J.; van Stevendaal, M.H.M.E.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Van, N.G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018, 19, 213–228. [Google Scholar]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci. 2019, 20, E840. [Google Scholar] [CrossRef]
- Venkataraman, G.; Mirza, M.K.; Eichenauer, D.A.; Diehl, V. Current status of prognostication in classical Hodgkin lymphoma. Br. J. Haematol. 2014, 165, 287–299. [Google Scholar] [CrossRef]
- Kuppers, R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Englund, A.; Molin, D.; Enblad, G.; Karlen, J.; Glimelius, I.; Ljungman, G.; Amini, R.M. The role of tumour-infiltrating eosinophils, mast cells and macrophages in Classical and Nodular Lymphocyte Predominant Hodgkin Lymphoma in children. Eur. J. Haematol. 2016, 97, 430–438. [Google Scholar] [CrossRef]
- Tan, K.L.; Scott, D.W.; Hong, F.; Kahl, B.S.; Fisher, R.I.; Bartlett, N.L.; Advani, R.H.; Buckstein, R.; Rimsza, L.M.; Connors, J.M.; et al. Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: A correlative study from the E2496 Intergroup trial. Blood 2012, 120, 3280–3287. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef]
- Vardhana, S.; Younes, A. The immune microenvironment in Hodgkin lymphoma: T cells, B cells, and immune checkpoints. Haematologica 2016, 101, 794–802. [Google Scholar] [CrossRef]
- Andersen, M.D.; Kamper, P.; Nielsen, P.S.; Bendix, K.; Riber-Hansen, R.; Steiniche, T.; Hamilton-Dutoit, S.; Clausen, M.; d’Amore, F. Tumour-associated mast cells in classical Hodgkin’s lymphoma: Correlation with histological subtype, other tumour-infiltrating inflammatory cell subsets and outcome. Eur. J. Haematol. 2016, 96, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Gholiha, A.R.; Hollander, P.; Hedstrom, G.; Sundstrom, C.; Molin, D.; Smedby, K.E.; Hjalgrim, H.; Glimelius, I.; Amini, R.M.; Enblad, G. High tumour plasma cell infiltration reflects an important microenvironmental component in classic Hodgkin lymphoma linked to presence of B-symptoms. Br. J. Haematol. 2019, 184, 192–201. [Google Scholar] [CrossRef]
- Aldinucci, D.; Lorenzon, D.; Olivo, K.; Rapana, B.; Gattei, V. Interactions between tissue fibroblasts in lymph nodes and Hodgkin/Reed-Sternberg cells. Leuk Lymphoma. 2004, 45, 1731–1739. [Google Scholar] [CrossRef]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Linke, F.; Harenberg, M.; Nietert, M.M.; Zaunig, S.; von, B.F.; Arlt, A.; Szczepanowski, M.; Weich, H.A.; Lutz, S.; Dullin, C.; et al. Microenvironmental interactions between endothelial and lymphoma cells: A role for the canonical WNT pathway in Hodgkin lymphoma. Leukemia 2017, 31, 361–372. [Google Scholar] [CrossRef]
- Liu, Y.; Sattarzadeh, A.; Diepstra, A.; Visser, L.; van den Berg, A. The microenvironment in classical Hodgkin lymphoma: An actively shaped and essential tumor component. Semin Cancer Biol. 2014, 24, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; Colombatti, A.; Carbone, A. The role of CD40/CD40L and interferon regulatory factor 4 in Hodgkin lymphoma microenvironment. Leuk Lymphoma. 2012, 53, 195–201. [Google Scholar] [CrossRef]
- Hartmann, S.; Jakobus, C.; Rengstl, B.; Doring, C.; Newrzela, S.; Brodt, H.R.; Wolf, T.; Hansmann, M.L. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells in HIV-related classical Hodgkin lymphoma. Mod. Pathol. 2013, 26, 648–657. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; de, P.P.; Zagonel, V.; Pinto, A. Expression of functional CD40 antigen on Reed-Sternberg cells and Hodgkin’s disease cell lines. Blood 1995, 85, 780–789. [Google Scholar] [PubMed]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef]
- Hanamoto, H.; Nakayama, T.; Miyazato, H.; Takegawa, S.; Hieshima, K.; Tatsumi, Y.; Kanamaru, A.; Yoshie, O. Expression of CCL28 by Reed-Sternberg cells defines a major subtype of classical Hodgkin’s disease with frequent infiltration of eosinophils and/or plasma cells. Am. J. Pathol. 2004, 164, 997–1006. [Google Scholar] [CrossRef]
- Fischer, M.; Juremalm, M.; Olsson, N.; Backlin, C.; Sundstrom, C.; Nilsson, K.; Enblad, G.; Nilsson, G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int. J. Cancer 2003, 107, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen 1 in Hodgkin’s lymphoma cells mediates Up-regulation of CCL20 and the migration of regulatory T cells. Am. J. Pathol 2008, 173, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, A.; Visser, L.; Poppema, S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am. J. Pathol 1999, 154, 1685–1691. [Google Scholar] [CrossRef]
- Niens, M.; Visser, L.; Nolte, I.M.; van der Steege, G.; Diepstra, A.; Cordano, P.; Jarrett, R.F.; Te Meerman, G.J.; Poppema, S.; van den Berg, A. Serum chemokine levels in Hodgkin lymphoma patients: Highly increased levels of CCL17 and CCL22. Br. J. Haematol. 2008, 140, 527–536. [Google Scholar] [CrossRef]
- Lamprecht, B.; Kreher, S.; Anagnostopoulos, I.; Johrens, K.; Monteleone, G.; Jundt, F.; Stein, H.; Janz, M.; Dorken, B.; Mathas, S. Aberrant expression of the Th2 cytokine IL-21 in Hodgkin lymphoma cells regulates STAT3 signaling and attracts Treg cells via regulation of MIP-3alpha. Blood 2008, 112, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Poppema, S.; van den Berg, A. Interaction between host T cells and Reed-Sternberg cells in Hodgkin lymphomas. Semin Cancer Biol. 2000, 10, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Olivo, K.; Lorenzon, D.; Poletto, D.; Gloghini, A.; Carbone, A.; Pinto, A. The role of interleukin-3 in classical Hodgkin’s disease. Leuk Lymphoma. 2005, 46, 303–311. [Google Scholar] [CrossRef]
- Pinto, A.; Aldinucci, D.; Gloghini, A.; Zagonel, V.; Degan, M.; Perin, V.; Todesco, M.; De, I.A.; Improta, S.; Sacco, C.; Gattei, V.; et al. The role of eosinophils in the pathobiology of Hodgkin’s disease. Ann. Oncol. 1997, 8 Suppl 2, 89–96. [Google Scholar] [CrossRef]
- Aldinucci, D.; Poletto, D.; Gloghini, A.; Nanni, P.; Degan, M.; Perin, T.; Ceolin, P.; Rossi, F.M.; Gattei, V.; Carbone, A.; et al. Expression of functional interleukin-3 receptors on Hodgkin and Reed-Sternberg cells. Am. J. Pathol. 2002, 160, 585–596. [Google Scholar] [CrossRef]
- Samoszuk, M.; Nansen, L. Detection of interleukin-5 messenger RNA in Reed-Sternberg cells of Hodgkin’s disease with eosinophilia. Blood 1990, 75, 13–16. [Google Scholar]
- Kapp, U.; Yeh, W.C.; Patterson, B.; Elia, A.J.; Kagi, D.; Ho, A.; Hessel, A.; Tipsword, M.; Williams, A.; Mirtsos, C.; et al. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J. Exp. Med. 1999, 189, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Foss, H.D.; Hummel, M.; Gottstein, S.; Ziemann, K.; Falini, B.; Herbst, H.; Stein, H. Frequent expression of IL-7 gene transcripts in tumor cells of classical Hodgkin’s disease. Am. J. Pathol. 1995, 146, 33–39. [Google Scholar] [PubMed]
- Cattaruzza, L.; Gloghini, A.; Olivo, K.; Di, F.R.; Lorenzon, D.; De, F.R.; Carbone, A.; Colombatti, A.; Pinto, A.; Aldinucci, D. Functional coexpression of Interleukin (IL)-7 and its receptor (IL-7R) on Hodgkin and Reed-Sternberg cells: Involvement of IL-7 in tumor cell growth and microenvironmental interactions of Hodgkin’s lymphoma. Int. J. Cancer. 2009, 125, 1092–1101. [Google Scholar] [CrossRef]
- Skinnider, B.F.; Elia, A.J.; Gascoyne, R.D.; Trumper, L.H.; von, B.F.; Kapp, U.; Patterson, B.; Snow, B.E.; Mak, T.W. Interleukin 13 and interleukin 13 receptor are frequently expressed by Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2001, 97, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, K.; Blumenthal-Barby, F.; Lamprecht, B.; Kochert, K.; Lenze, D.; Hummel, M.; Mathas, S.; Dorken, B.; Janz, M. The IL-15 cytokine system provides growth and survival signals in Hodgkin lymphoma and enhances the inflammatory phenotype of HRS cells. Leukemia 2015, 29, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Fiumara, P.; Snell, V.; Li, Y.; Mukhopadhyay, A.; Younes, M.; Gillenwater, A.M.; Cabanillas, F.; Aggarwal, B.B.; Younes, A. Functional expression of receptor activator of nuclear factor kappaB in Hodgkin disease cell lines. Blood 2001, 98, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.W.; et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007, 109, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, J.; Went, P.; Matthes, T.; Dirnhofer, S.; Donze, O.; Mhawech-Fauceglia, P.; Myit, S.; Huard, B. Paracrine promotion of tumor development by the TNF ligand APRIL in Hodgkin’s Disease. Leukemia 2007, 21, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Sugihara, M.; Suzumiya, J.; Haraoka, S.; Kanda, M.; Shimazaki, K.; Katoh, K.; Kumagawa, M.; Kikuchi, M. Basic fibroblast growth factor and fibrosis in Hodgkin’s disease. Pathol. Res. Pract. 1999, 195, 149–155. [Google Scholar] [CrossRef]
- Celegato, M.; Borghese, C.; Casagrande, N.; Mongiat, M.; Kahle, X.U.; Paulitti, A.; Spina, M.; Colombatti, A.; Aldinucci, D. Preclinical activity of the repurposed drug Auranofin in classical Hodgkin lymphoma. Blood 2015, 126, 1394–1397. [Google Scholar] [CrossRef]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 7, 1154–1167. [Google Scholar] [CrossRef]
- Klein, S.; Jucker, M.; Diehl, V.; Tesch, H. Production of multiple cytokines by Hodgkin’s disease derived cell lines. Hematol. Oncol. 1992, 10, 319–329. [Google Scholar] [CrossRef]
- Hong, I.S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed]
- Jundt, F.; Anagnostopoulos, I.; Forster, R.; Mathas, S.; Stein, H.; Dorken, B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 2002, 99, 3398–3403. [Google Scholar] [CrossRef] [PubMed]
- Foss, H.D.; Herbst, H.; Oelmann, E.; Samol, J.; Grebe, M.; Blankenstein, T.; Matthes, J.; Qin, Z.H.; Falini, B.; Pileri, S. Lymphotoxin, tumour necrosis factor and interleukin-6 gene transcripts are present in Hodgkin and Reed-Sternberg cells of most Hodgkin’s disease cases. Br. J. Haematol. 1993, 84, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.L.; Hsu, S.M. Production of tumor necrosis factor-alpha and lymphotoxin by cells of Hodgkin’s neoplastic cell lines HDLM-1 and KM-H2. Am. J. Pathol. 1989, 135, 735–745. [Google Scholar]
- Fhu, C.W.; Graham, A.M.; Yap, C.T.; Al-Salam, S.; Castella, A.; Chong, S.M.; Lim, Y.C. Reed-Sternberg cell-derived lymphotoxin-alpha activates endothelial cells to enhance T-cell recruitment in classical Hodgkin lymphoma. Blood 2014, 124, 2973–2982. [Google Scholar] [CrossRef]
- Moreau, A.; Praloran, V.; Berrada, L.; Coupey, L.; Gaillard, F. Immunohistochemical detection of cells positive for colony-stimulating factor 1 in lymph nodes from reactive lymphadenitis, and Hodgkin’s disease. Leukemia. 1992, 6, 126–130. [Google Scholar] [PubMed]
- Paietta, E.; Racevskis, J.; Stanley, E.R.; Andreeff, M.; Papenhausen, P.; Wiernik, P.H. Expression of the macrophage growth factor, CSF-1 and its receptor c-fms by a Hodgkin’s disease-derived cell line and its variants. Cancer Res. 1990, 50, 2049–2055. [Google Scholar] [PubMed]
- Tudor, C.S.; Bruns, H.; Daniel, C.; Distel, L.V.; Hartmann, A.; Gerbitz, A.; Buettner, M.J. Macrophages and dendritic cells as actors in the immune reaction of classical Hodgkin lymphoma. PLoS One. 2014, 9, e114345. [Google Scholar] [CrossRef]
- Kadin, M.E.; Agnarsson, B.A.; Ellingsworth, L.R.; Newcom, S.R. Immunohistochemical evidence of a role for transforming growth factor beta in the pathogenesis of nodular sclerosing Hodgkin’s disease. Am. J. Pathol. 1990, 136, 1209–1214. [Google Scholar] [PubMed]
- Hsu, S.M.; Lin, J.; Xie, S.S.; Hsu, P.L.; Rich, S. Abundant expression of transforming growth factor-beta 1 and -beta 2 by Hodgkin’s Reed-Sternberg cells and by reactive T lymphocytes in Hodgkin’s disease. Hum. Pathol. 1993, 24, 249–255. [Google Scholar] [CrossRef]
- Kadin, M.; Butmarc, J.; Elovic, A.; Wong, D. Eosinophils are the major source of transforming growth factor-beta 1 in nodular sclerosing Hodgkin’s disease. Am. J. Pathol. 1993, 142, 11–16. [Google Scholar] [PubMed]
- Skinnider, B.F.; Mak, T.W. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002, 99, 4283–4297. [Google Scholar] [CrossRef] [PubMed]
- Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Emmerich, F.; Muller, G.; Foss, H.D.; Royer, H.D.; Stein, H.; Dorken, B. Hodgkin/Reed-Sternberg cells induce fibroblasts to secrete eotaxin, a potent chemoattractant for T cells and eosinophils. Blood 1999, 94, 2065–2071. [Google Scholar] [PubMed]
- Doussis-Anagnostopoulou, I.A.; Talks, K.L.; Turley, H.; Debnam, P.; Tan, D.C.; Mariatos, G.; Gorgoulis, V.; Kittas, C.; Gatter, K.C. Vascular endothelial growth factor (VEGF) is expressed by neoplastic Hodgkin-Reed-Sternberg cells in Hodgkin’s disease. J. Pathol. 2002, 197, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Marinaccio, C.; Nico, B.; Maiorano, E.; Specchia, G.; Ribatti, D. Insights in Hodgkin Lymphoma angiogenesis. Leuk. Res. 2014, 38, 857–861. [Google Scholar] [CrossRef]
- Buri, C.; Korner, M.; Scharli, P.; Cefai, D.; Uguccioni, M.; Mueller, C.; Laissue, J.A.; Mazzucchelli, L. CC chemokines and the receptors CCR3 and CCR5 are differentially expressed in the nonneoplastic leukocytic infiltrates of Hodgkin disease. Blood 2001, 97, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Maggio, E.; van den Berg, A.; Diepstra, A.; Kluiver, J.; Visser, L.; Poppema, S. Chemokines, cytokines and their receptors in Hodgkin’s lymphoma cell lines and tissues. Ann. Oncol. 2002, 13 Suppl 1, 52–56. [Google Scholar] [CrossRef]
- Meadows, S.A.; Vega, F.; Kashishian, A.; Johnson, D.; Diehl, V.; Miller, L.L.; Younes, A.; Lannutti, B.J. PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood 2012, 119, 1897–1900. [Google Scholar] [CrossRef]
- Teruya-Feldstein, J.; Jaffe, E.S.; Burd, P.R.; Kingma, D.W.; Setsuda, J.E.; Tosato, G. Differential chemokine expression in tissues involved by Hodgkin’s disease: Direct correlation of eotaxin expression and tissue eosinophilia. Blood 1999, 93, 2463–2470. [Google Scholar]
- Buglio, D.; Georgakis, G.V.; Hanabuchi, S.; Arima, K.; Khaskhely, N.M.; Liu, Y.J.; Younes, A. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood 2008, 112, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Peh, S.C.; Kim, L.H.; Poppema, S. TARC, a CC chemokine, is frequently expressed in classic Hodgkin’s lymphoma but not in NLP Hodgkin’s lymphoma, T-cell-rich B-cell lymphoma, and most cases of anaplastic large cell lymphoma. Am. J. Surg. Pathol. 2001, 25, 925–929. [Google Scholar] [CrossRef]
- Hedvat, C.V.; Jaffe, E.S.; Qin, J.; Filippa, D.A.; Cordon-Cardo, C.; Tosato, G.; Nimer, S.D.; Teruya-Feldstein, J. Macrophage-derived chemokine expression in classical Hodgkin’s lymphoma: Application of tissue microarrays. Mod. Pathol. 2001, 14, 1270–1276. [Google Scholar] [CrossRef][Green Version]
- Di, S.A.; De, A.B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar]
- Aldinucci, D.; Rapana, B.; Olivo, K.; Lorenzon, D.; Gloghini, A.; Colombatti, A.; Carbone, A. IRF4 is modulated by CD40L and by apoptotic and anti-proliferative signals in Hodgkin lymphoma. Br. J. Haematol. 2010, 148, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Jundt, F.; Probsting, K.S.; Anagnostopoulos, I.; Muehlinghaus, G.; Chatterjee, M.; Mathas, S.; Bargou, R.C.; Manz, R.; Stein, H.; Dorken, B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004, 103, 3511–3515. [Google Scholar] [CrossRef]
- Rajendran, S.; Ho, W.T.; Schwarz, H. CD137 signaling in Hodgkin and Reed-Sternberg cell lines induces IL-13 secretion, immune deviation and enhanced growth. Oncoimmunology 2016, 5, e1160188. [Google Scholar] [CrossRef] [PubMed]
- Cader, F.Z.; Vockerodt, M.; Bose, S.; Nagy, E.; Brundler, M.A.; Kearns, P.; Murray, P.G. The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood 2013, 122, 4237–4245. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular pathogenesis of Hodgkin’s lymphoma: Increasing evidence of the importance of the microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef]
- Glimelius, I.; Edstrom, A.; Fischer, M.; Nilsson, G.; Sundstrom, C.; Molin, D.; Amini, R.M.; Enblad, G. Angiogenesis and mast cells in Hodgkin lymphoma. Leukemia 2005, 19, 2360–2362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; Armand, P.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4(+) regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Ansell, S.M. Targeting immune checkpoints in lymphoma. Curr. Opin. Hematol. 2015, 22, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Ansell, S.M. Novel agents in the treatment of Hodgkin lymphoma: Biological basis and clinical results. Semin Hematol. 2016, 53, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; Moskowitz, C.H. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef]
- Koh, Y.W.; Park, C.S.; Yoon, D.H.; Suh, C.; Huh, J. CD163 expression was associated with angiogenesis and shortened survival in patients with uniformly treated classical Hodgkin lymphoma. PLoS One 2014, 9, e87066. [Google Scholar] [CrossRef]
- Crane, G.M.; Samols, M.A.; Morsberger, L.A.; Yonescu, R.; Thiess, M.L.; Batista, D.A.; Ning, Y.; Burns, K.H.; Vuica-Ross, M.; Borowitz, M.J.; et al. Tumor-Infiltrating Macrophages in Post-Transplant, Relapsed Classical Hodgkin Lymphoma Are Donor-Derived. PLoS One 2016, 11, e0163559. [Google Scholar] [CrossRef]
- Gotti, M.; Nicola, M.; Lucioni, M.; Fiaccadori, V.; Ferretti, V.; Sciarra, R.; Costanza, M.; Bono, E.; Molo, S.; Maffi, A.; et al. Independent prognostic impact of tumour-infiltrating macrophages in early-stage Hodgkin’s lymphoma. Hematol Oncol. 2017, 35, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; Hodi, F.S.; Liu, X.S.; Shipp, M.A.; Rodig, S.J. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; Cross, D.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Merino, L.; Lejeune, M.; Nogales, F.E.; Henao, C.F.; Grueso, L.A.; Illescas, V.A.; Pulla, M.P.; Callau, C.; Alvaro, T. Role of immune escape mechanisms in Hodgkin’s lymphoma development and progression: A whole new world with therapeutic implications. Clin. Dev. Immunol. 2012, 2012, 756353. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Vetro, C.; Caocci, G.; Greco, M.; Parrinello, N.L.; Di, R.F.; La, N.G. Immunological deregulation in classic hodgkin lymphoma. Mediterr. J. Hematol Infect. Dis. 2014, 6, e2014039. [Google Scholar] [PubMed]
- Juszczynski, P.; Ouyang, J.; Monti, S.; Rodig, S.J.; Takeyama, K.; Abramson, J.; Chen, W.; Kutok, J.L.; Rabinovich, G.A.; Shipp, M.A. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 13134–13139. [Google Scholar] [CrossRef] [PubMed]
- Oelmann, E.; Herbst, H.; Zuhlsdorf, M.; Albrecht, O.; Nolte, A.; Schmitmann, C.; Manzke, O.; Diehl, V.; Stein, H.; Berdel, W.E. Tissue inhibitor of metalloproteinases 1 is an autocrine and paracrine survival factor, with additional immune-regulatory functions, expressed by Hodgkin/Reed-Sternberg cells. Blood 2002, 99, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Driesen, J.; Classen, S.; Riley, J.L.; Debey, S.; Beyer, M.; Popov, A.; Zander, T.; Schultze, J.L. Prostaglandin E2 impairs CD4+ T cell activation by inhibition of lck: implications in Hodgkin’s lymphoma. Cancer Res. 2006, 66, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Wein, F.; Weniger, M.A.; Hoing, B.; Arnolds, J.; Huttmann, A.; Hansmann, M.L.; Hartmann, S.; Kuppers, R. Complex Immune Evasion Strategies in Classical Hodgkin Lymphoma. Cancer Immunol. Res. 2017, 5, 1122–1132. [Google Scholar] [CrossRef]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Johrens-Leder, K.; Vornlocher, H.P.; Bommert, K.; et al. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004, 199, 1041–1052. [Google Scholar] [CrossRef]
- Poppema, S. Immunobiology and pathophysiology of Hodgkin lymphomas. Hematology Am. Soc. Hematol Educ. Program. 2005, 231–238. [Google Scholar] [CrossRef]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef]
- Caocci, G.; Greco, M.; Fanni, D.; Senes, G.; Littera, R.; Lai, S.; Risso, P.; Carcassi, C.; Faa, G.; La, N.G. HLA-G expression and role in advanced-stage classical Hodgkin lymphoma. Eur. J. Histochem. 2016, 60, 2606. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.T.; Pang, W.L.; Chong, S.M.; Castella, A.; Al-Salam, S.; Tan, T.E.; Moh, M.C.; Koh, L.K.; Gan, S.U.; Cheng, C.K.; Schwarz, H. Expression of CD137 on Hodgkin and Reed-Sternberg cells inhibits T-cell activation by eliminating CD137 ligand expression. Cancer Res. 2013, 73, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Nishikori, M.; Kitawaki, T.; Sakai, T.; Hishizawa, M.; Tashima, M.; Kondo, T.; Ohmori, K.; Kurata, M.; Hayashi, T.; et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008, 111, 3220–3224. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Masaki, A.; Ishida, T.; Maeda, Y.; Ito, A.; Suzuki, S.; Narita, T.; Kinoshita, S.; Takino, H.; Yoshida, T.; Ri, M.; et al. Clinical significance of tryptophan catabolism in Hodgkin lymphoma. Cancer Sci. 2018, 109, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Yun, J.Y.; Jeon, Y.K.; Kim, S.H.; Park, G.; Huh, J.R.; Oh, S.; Kim, J.E. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: A retrospective cohort study. BMC Cancer. 2014, 14, 335. [Google Scholar] [CrossRef]
- Zeiser, R.; Robson, S.C.; Vaikunthanathan, T.; Dworak, M.; Burnstock, G. Unlocking the Potential of Purinergic Signaling in Transplantation. Am. J. Transplant. 2016, 16, 2781–2794. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, Y.; Visser, L.; Blokzijl, T.; Harms, G.; Atayar, C.; Poppema, S.; van den Berg, A. The CD4+CD26- T-cell population in classical Hodgkin’s lymphoma displays a distinctive regulatory T-cell profile. Lab. Invest. 2008, 88, 482–490. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.L.; Careddu, G.; Serio, S.; Consonni, F.M.; Maeda, A.; Viswanadha, S.; Vakkalanka, S.; Castagna, L.; Santoro, A.; Allavena, P.; Sica, A.; et al. Targeting cancer cells and tumor microenvironment in preclinical and clinical models of Hodgkin lymphoma using the dual PI3Kdelta/gamma inhibitor RP6530. Clin. Cancer Res. 2019, 25, 1098–1112. [Google Scholar]
- Iqbal, M.A.; Gupta, V.; Gopinath, P.; Mazurek, S.; Bamezai, R.N. Pyruvate kinase M2 and cancer: An updated assessment. FEBS Lett. 2014, 588, 2685–2692. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Johnson, T.S. Treg Destabilization and Reprogramming: Implications for Cancer Immunotherapy. Cancer Res. 2018, 78, 5191–5199. [Google Scholar] [CrossRef]
- Deng, G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018, 7, 81–87. [Google Scholar]
- Aldinucci, D.; Pinto, A.; Gloghini, A.; Carbone, A. Chemokine receptors as therapeutic tools in Hodgkin lymphoma: CCR4 and beyond. Blood 2010, 115, 746–747. [Google Scholar] [CrossRef][Green Version]
- Tanijiri, T.; Shimizu, T.; Uehira, K.; Yokoi, T.; Amuro, H.; Sugimoto, H.; Torii, Y.; Tajima, K.; Ito, T.; Amakawa, R.; et al. Hodgkin’s reed-sternberg cell line (KM-H2) promotes a bidirectional differentiation of CD4+CD25+Foxp3+ T cells and CD4+ cytotoxic T lymphocytes from CD4+ naive T cells. J. Leukoc. Biol. 2007, 82, 576–584. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Glod, J.W.; Banerjee, D. Mesenchymal stem cells: Flip side of the coin. Cancer Res. 2009, 69, 1255–1258. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R. Stress immunity in lymphomas: Mesenchymal cells as a target of therapy. Front. Biosci (Landmark Ed.) 2014, 19, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tableros, V.; Gomez, Y.; Camussi, G.; Brizzi, M.F. Extracellular Vesicles: New Players in Lymphomas. Int. J. Mol. Sci. 2018, 20, 41. [Google Scholar] [CrossRef]
- Hansen, H.P.; Engels, H.M.; Dams, M.; Paes Leme, A.F.; Pauletti, B.A.; Simhadri, V.L.; Durkop, H.; Reiners, K.S.; Barnert, S.; Engert, A.; Schubert, R.; Quondamatteo, F.; Hallek, M.; Pogge von, S.E. Protrusion-guided extracellular vesicles mediate CD30 trans-signalling in the microenvironment of Hodgkin’s lymphoma. J. Pathol. 2014, 232, 405–414. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel) 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, B.; Bosl, T.; Reiners, K.S.; Barnert, S.; Schubert, R.; Shatnyeva, O.; Zigrino, P.; Engert, A.; Hansen, H.P.; von Strandmann, E.P. Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front. Immunol. 2018, 9, 1358. [Google Scholar] [CrossRef]
- Tosetti, F.; Vene, R.; Camodeca, C.; Nuti, E.; Rossello, A.; D’Arrigo, C.; Galante, D.; Ferrari, N.; Poggi, A.; Zocchi, M.R. Specific ADAM10 inhibitors localize in exosome-like vesicles released by Hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology. 2018, 7, e1421889. [Google Scholar] [CrossRef]
- Ansell, S.M. Brentuximab vedotin. Blood 2014, 124, 3197–3200. [Google Scholar] [CrossRef]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Cirillo, M.; Reinke, S.; Klapper, W.; Borchmann, S. The translational science of hodgkin lymphoma. Br. J. Haematol. 2019, 184, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.; Patel, S.P.; Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 2017, 14, 203–220. [Google Scholar] [CrossRef]
- Greaves, P.; Clear, A.; Owen, A.; Iqbal, S.; Lee, A.; Matthews, J.; Wilson, A.; Calaminici, M.; Gribben, J.G. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood 2013, 122, 2856–2863. [Google Scholar] [CrossRef] [PubMed]
- Von, K.G.; Younes, A. Novel therapeutic agents for relapsed classical Hodgkin lymphoma. Br. J. Haematol. 2019, 184, 105–112. [Google Scholar]
- Pericart, S.; Tosolini, M.; Gravelle, P.; Rossi, C.; Traverse-Glehen, A.; Amara, N.; Franchet, C.; Martin, E.; Bezombes, C.; Laurent, G.; et al. Profiling Immune Escape in Hodgkin’s and Diffuse large B-Cell Lymphomas Using the Transcriptome and Immunostaining. Cancers (Basel) 2018, 10, 415. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Boulakirba, S.; Pfeifer, A.; Mhaidly, R.; Obba, S.; Goulard, M.; Schmitt, T.; Chaintreuil, P.; Calleja, A.; Furstoss, N.; Orange, F.; et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018, 8, 256–18433. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Diepstra, A.; Lee, T.; Chan, F.C.; Farinha, P.; Tan, K.; Telenius, A.; Barclay, L.; Shah, S.P.; Connors, J.M.; van den Berg, A.; Gascoyne, R.D. Gene expression profiling of microdissected Hodgkin Reed-Sternberg cells correlates with treatment outcome in classical Hodgkin lymphoma. Blood 2012, 120, 3530–3540. [Google Scholar] [CrossRef]
- Von, T.B.; Morschhauser, F.; Ribrag, V.; Topp, M.S.; Chien, C.; Seetharam, S.; Aquino, R.; Kotoulek, S.; de Boer, C.J.; Engert, A. An Open-Label, Multicenter, Phase I/II Study of JNJ-40346527, a CSF-1R Inhibitor, in Patients with Relapsed or Refractory Hodgkin Lymphoma. Clin. Cancer Res. 2015, 21, 1843–1850. [Google Scholar]
- Moskowitz, C.H.; Younes, A.; De Vos, S.; Bociek, G.; Gordon, L.I.; Witzig, T.E.; Gascoyne, R.D.; West, B.; Nolop, K.; Steidl, C. CSF1R Inhibition by PLX3397 in Patients with Relapsed or Refractory Hodgkin Lymphoma: Results From a Phase 2 Single Agent Clinical Tria. Blood 2012, 120, 1638. [Google Scholar]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015, 125, 4024–4031. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Lemos, H.; Huang, L.; Prendergast, G.C.; Mellor, A.L. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer. 2019, 19, 162–175. [Google Scholar] [CrossRef]
- Davar, D.; Bahary, N. Modulating Tumor Immunology by Inhibiting Indoleamine 2,3-Dioxygenase (IDO): Recent Developments and First Clinical Experiences. Target. Oncol. 2018, 13, 125–140. [Google Scholar] [CrossRef]
- Abel, S.; van der Ryst, E.; Rosario, M.C.; Ridgway, C.E.; Medhurst, C.G.; Taylor-Worth, R.J.; Muirhead, G.J. Assessment of the pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist, in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65 Suppl 1, 5–18. [Google Scholar] [CrossRef]
- Genebat, M.; Ruiz-Mateos, E.; Pulido, I.; Gonzalez-Serna, A.; Garcia-Perganeda, A.; Mendez, G.; Romero-Sanchez, M.C.; Ferrando-Martinez, S.; Leal, M. Long-term immunovirogical effect and tolerability of a maraviroc-containing regimen in routine clinical practice. Curr. HIV Res. 2010, 8, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Weir, S.J.; DeGennaro, L.J.; Austin, C.P. Repurposing approved and abandoned drugs for the treatment and prevention of cancer through public-private partnership. Cancer Res. 2012, 72, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velazquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology 2016, 5, e1150398. [Google Scholar] [CrossRef]
- Tanabe, Y.; Sasaki, S.; Mukaida, N.; Baba, T. Blockade of the chemokine receptor, CCR5, reduces the growth of orthotopically injected colon cancer cells via limiting cancer-associated fibroblast accumulation. Oncotarget 2016, 7, 48335–48345. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell. 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; Flowers, C.R.; Martin, P.; Ansell, S.M.; Abella-Dominicis, E.; Koh, B.; Ye, W.; Barr, P.M.; Salles, G.A.; Friedberg, J.W. Idelalisib is effective in patients with high-risk follicular lymphoma and early relapse after initial chemoimmunotherapy. Blood 2017, 129, 3037–3039. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Harrington, B.K.; Hu, E.Y.; Greene, J.T.; Lehman, A.M.; Tran, M.; Wasmuth, R.L.; Long, M.; Muthusamy, N.; Brown, J.R.; Johnson, A.J.; Byrd, J.C. PI3K p110delta inactivation antagonizes chronic lymphocytic leukemia and reverses T cell immune suppression. J. Clin. Invest. 2019, 129, 122–136. [Google Scholar] [CrossRef]
- De, H.O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; Douglas, M.; Tibbitts, T.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar]
- Locatelli, S.L.; Careddu, G.; Inghirami, G.; Castagna, L.; Sportelli, P.; Santoro, A.; Carlo-Stella, C. The novel PI3K-delta inhibitor TGR-1202 enhances Brentuximab Vedotin-induced Hodgkin lymphoma cell death via mitotic arrest. Leukemia 2016, 30, 2402–2405. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Fanale, M.A.; Moskowitz, C.H.; Shustov, A.R.; Mitra, S.; Ye, W.; Younes, A.; Moskowitz, A.J. Phase II study of idelalisib, a selective inhibitor of PI3Kdelta, for relapsed/refractory classical Hodgkin lymphoma. Ann. Oncol. 2017, 28, 1057–1063. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef]
- Fromm, J.R. Flow cytometric analysis of CD123 is useful for immunophenotyping classical Hodgkin lymphoma. Cytometry B Clin. Cytom. 2011, 80, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Mannarino, L.; Paracchini, L.; Craparotta, I.; Romano, M.; Marchini, S.; Gatta, R.; Erba, E.; Clivio, L.; Romualdi, C.; D’Incalci, M.; et al. A systems biology approach to investigate the mechanism of action of trabectedin in a model of myelomonocytic leukemia. Pharmacogenom. J. 2016, 18, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Della Porta, M.G.; Galli, A.; Panini, N.; Licandro, S.A.; Bello, E.; Craparotta, I.; Rosti, V.; Bonetti, E.; Tancredi, R.; Rossi, M.; Mannarino, L.; et al. Antitumour activity of trabectedin in myelodysplastic/myeloproliferative neoplasms. Br. J. Cancer. 2017, 116, 335–343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spriano, F.; Chung, E.Y.; Panini, N.; Cascione, L.; Rinaldi, A.; Erba, E.; Stathis, A.; D’Incalci, M.; Bertoni, F.; Gatta, R. Trabectedin is a novel chemotherapy agent for diffuse large B cell lymphoma. Br. J. Haematol. 2019, 184, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Kaur, V.; Kumar, M.; Kaur, P.; Murthy, R.S.; Rawal, R.K. The critical role of bisphosphonates to target bone cancer metastasis: An overview. J. Drug Target. 2015, 23, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; De Luca, A.; Lamura, L.; Normanno, N. Zoledronic acid blocks the interaction between mesenchymal stem cells and breast cancer cells: Implications for adjuvant therapy of breast cancer. Ann. Oncol. 2012, 23, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Borghese, C.; Casagrande, N.; Pivetta, E.; Colombatti, A.; Boccellino, M.; Amler, E.; Normanno, N.; Caraglia, M.; De Rosa, G.; Aldinucci, D. Self-assembling nanoparticles encapsulating zoledronic acid inhibit mesenchymal stromal cells differentiation, migration and secretion of proangiogenic factors and their interactions with prostate cancer cells. Oncotarget 2017, 8, 42926–42938. [Google Scholar] [CrossRef]
- Comito, G.; Segura, C.P.; Taddei, M.L.; Lanciotti, M.; Serni, S.; Morandi, A.; Chiarugi, P.; Giannoni, E. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget 2017, 8, 118–132. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cytokine | Expression by HRS (Tissue or Cell Lines) | Expression (TME) and Induction in Normal Cells | Function in cHL |
|---|---|---|---|
| IL-3 | Absent | Expressed by T cells and eosinophils [45] Induced in T cells by HRS cells [46] | Upregulation of CD40L and CD30L in eosinophils [47]; proliferation of eosinophils and mast cells [46]; proliferation of HRS cells [48] |
| IL-5 | Tissue [49] Cell lines [50] | Expressed by T cells [45] | Eosinophil recruitment [23]; upregulation of CD40L and CD30L in eosinophils [47] |
| IL-7 | Tissue [51] Cell lines [52] | Expressed by T cells [45] and HL fibroblasts [52] | Growth factor for Treg cells; proliferation of HRS cells; IL-7 stimulates IL-6 secretion by HL fibroblasts [52] |
| IL-13 | Tissue [53] Cell lines [53] | Rarely expressed by small lymphocytes [53] | Growth factor for fibroblasts [30]; autocrine growth factor for HRS cells [53] |
| IL-15 | Tissue [54] Cell lines [54,55] | Expressed by monocytes, dendritic cells, endothelial cells [54] | Proliferation, survival, and apoptosis resistance of HRS cells [54] |
| APRIL | Tissue [56] HL cell lines [56] | Neutrophils [56,57] | Proliferation of HRS cells [56,57] |
| FGF-2 | Tissue [58] Cell lines [59] | Expressed by stromal cells and histiocytes [58] | Growth factor for fibroblasts [30] and MSCs [40], endothelial cell tubulogenesis [32,59] |
| GM-CSF | Tissue [60] Cell lines [60,61] | Likely in activated T cells, B cells, macrophages, mast cells, endothelial cells and fibroblasts [62] | Recruitment of eosinophils [23]; up-regulation of CD40L and CD30L in eosinophils [47]; M2-TAM differentiation [60] |
| Jagged-1 | Tissue [63] Cell lines [59] | Endothelial cells, smooth muscle cells and epithelioid cells [63] | Proliferation and survival of HRS [63] |
| LT-α | Tissue [64] Cell lines [65] | Extracellular stroma [66] | Activates endothelial cells to enhance T cell recruitment [66] |
| M-CSF | Tissue [67] Cell lines [40,60,68] | Endothelial cells and fibroblasts [67] | Recruitment and proliferation of monocytes; differentiation of M2-TAM [40,69] |
| TGF-β | Tissue [70] Cell lines [59] | T lymphocytes; [71] eosinophils [72] Induced in monocytes by HRS [40] | Growth factor for fibroblasts [30] and MSCs [40]; endothelial cell tubulogenesis [32,59] |
| TNF-α | Tissue [64] Cell lines [59] | Lymphocytes and macrophages [73] | Growth factor for fibroblasts [30] and MSCs [40]; induction of eotaxin secretion by fibroblasts [74] |
| VEGF | Tissue [75] Cell lines [32,59] | Macrophages and lymphocytes [75,76] | Endothelial cell tubulogenesis [32,59] |
| CCL3 (MIP-1α) CCL4 (MIP-1β) | Low levels or absent | Macrophages [77] Increased in monocytes by HRS [40] | Proliferation of HRS [40] |
| CCL5 (RANTES) | Tissue [40,78] Cell lines [37,39,78] | T cells and B cells [77] Induced in MSCs [40] and fibroblasts [37] by HRS cells, or increased by cultivation of HRS cells with fibroblasts [79] | Recruitment of monocytes/macrophages and MSCs [40], eosinophils and T cells [37], and mast cells [39]; proliferation of HRS cells [37,40] |
| CCL11 (Eotaxin) | absent | Fibroblasts and some macrophages [45,74] and smooth muscle cells [80] Induced in fibroblasts by TNF-α secreted by HRS cells [74] | Recruitment of eosinophils and T cells by tumor fibroblasts [74] |
| CCL17 (TARC) | Tissue [42] Cell lines [42,59,81] | Occasional in macrophages [82] Induced in monocytes by HRS cells [40] | Recruitment of T cells and Tregs [42] |
| CCL20 (MIP-3α) | Tissue [41] Cell lines [41] | Some neutrophils [41] | Recruitment of Tregs [41] |
| CCL22 (MDC) | Tissue [43,83] Cell lines [78,80,84] | Rare in histiocytes and endothelial cells (weak cytoplasmic staining) [83] | Recruitment of Th2 and Tregs [43] |
| CCL28 (MEC) | Tissue [38] Cell lines [38] | Occasionally in TME [38] | Recruitment of eosinophils [38] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20, 2416. https://doi.org/10.3390/ijms20102416
Aldinucci D, Borghese C, Casagrande N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. International Journal of Molecular Sciences. 2019; 20(10):2416. https://doi.org/10.3390/ijms20102416
Chicago/Turabian StyleAldinucci, Donatella, Cinzia Borghese, and Naike Casagrande. 2019. "Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It" International Journal of Molecular Sciences 20, no. 10: 2416. https://doi.org/10.3390/ijms20102416
APA StyleAldinucci, D., Borghese, C., & Casagrande, N. (2019). Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. International Journal of Molecular Sciences, 20(10), 2416. https://doi.org/10.3390/ijms20102416