Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System


 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Zebrafish Resources to Mimic Human Diseases
3. Zebrafish Resources to Explore Mitochondrial Physiology
4. Modeling Mitochondrial Defects in Zebrafish
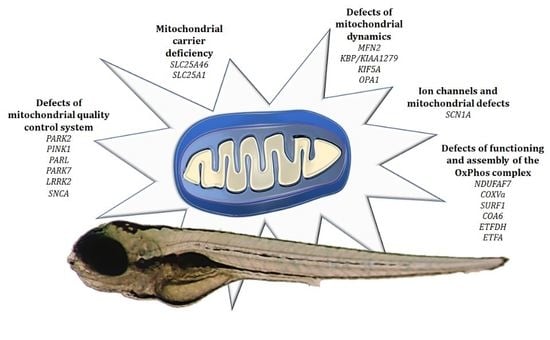
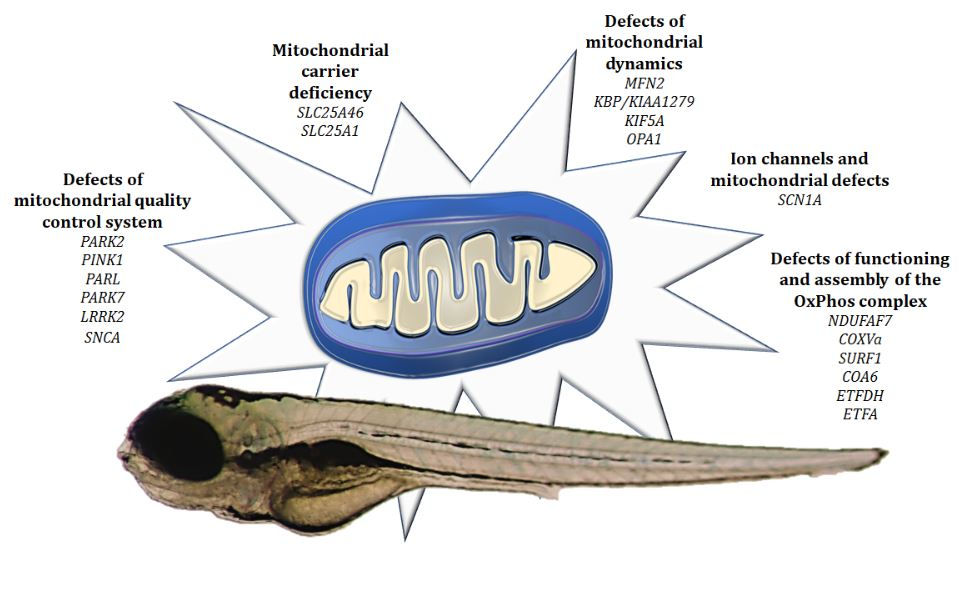
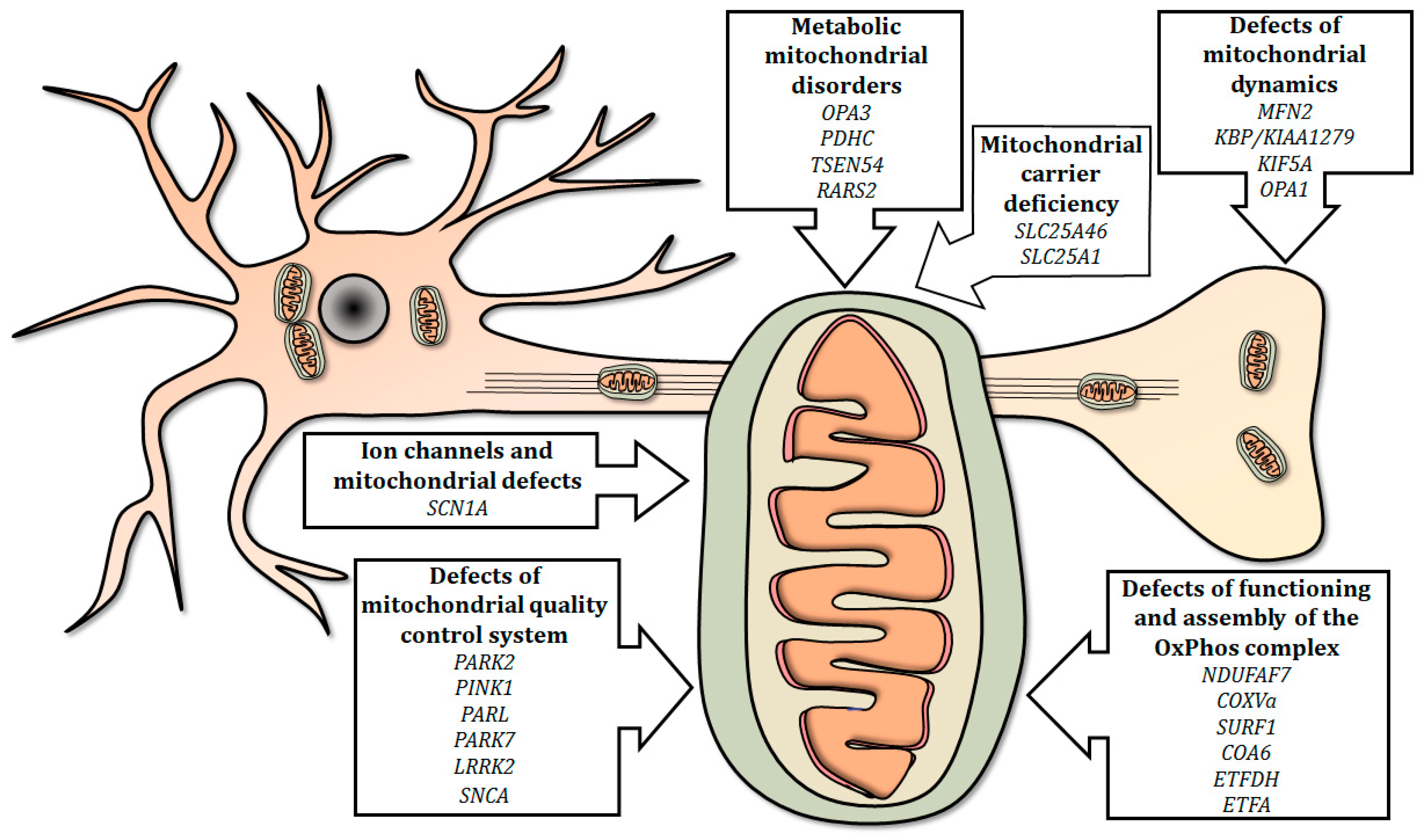
4.1. Defects of Functioning and Assembly of the OxPhos Complex
4.2. Ion Channels and Mitochondrial Defects
4.3. Defects of Mitochondrial Quality Control System
4.4. Defects of Mitochondrial Dynamics
4.5. Mitochondrial Carrier Deficiency
4.6. Metabolic Mitochondrial Disorders
5. Zebrafish as In Vivo Model to Test New Compounds in Mitochondrial Related Neurological Disorders
6. Materials and Methods
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| acetyl-CoA | Acetyl form of coenzyme A |
| ADOA | Autosomal dominant optic atrophy |
| Cas9 | CRISPR-associated protein 9 |
| CMT2 | Charcot-Marie-Tooth Type 2 |
| CNS | Central nervous system |
| COA | COX assembly factor |
| CoQ | Coenzyme Q |
| COX | Cytochrome c oxidase |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cys | Cysteine |
| cyt c | cytochrome c |
| DNP | 2-4 dinitrophenol |
| DOA | Dominant optic atrophy |
| DS | Dravet syndrome |
| EC | Enzyme complexes |
| ENU | N-ethyl-N-nitrosourea |
| Hcy | Homocysteine |
| MADD | Multiple acyl-CoA dehydrogenase deficiency |
| MCS | Mitochondrial carriers |
| MO | Morpholino oligonucleotide |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methylmethyl-4-phenylphenyl-1,2,3,6-tetrahydropyridinetetrahydropyridine |
| mRNA | Messenger RNA |
| mtDNA | Mitochondrial DNA |
| nDNA | Nuclear DNA |
| NGS | Next-generation sequencing |
| OxPhos | Oxidative phosphorylation |
| PBA | 4-phenylbutyrate |
| PCH | Pontocerebellar hypoplasia |
| PD | Parkinson’s disease |
| PDHC | Pyruvate dehydrogenase complex |
| pLL | Posterior lateral line |
| PNS | Peripheral nervous system |
| ROS | Reactive oxygen species |
| rRNA | Ribosomal RNA |
| TALEN | Transcription activator-like effector |
| tRNA | Transfer RNA |
References
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Lill, R. The role of mitochondria in cellular iron-sulfur protein biogenesis: Mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Beart, P.M.; Shin, Y.S.; Chen, M.J.; Cheung, N.S.; Nagley, P. Oxidative stress: Emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers Dis. 2010, 20, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays. Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Zeviani, M. Mitochondrial diseases: A cross-talk between mitochondrial and nuclear genomes. Adv. Exp. Med. Biol. 2009, 652, 69–84. [Google Scholar] [PubMed]
- Farrar, G.J.; Chadderton, N.; Kenna, P.F.; Millington-Ward, S. Mitochondrial disorders: Aetiologies, models systems, and candidate therapies. Trends Genet. 2013, 29, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef]
- Busch, K.B.; Kowald, A.; Spelbrink, J.N. Quality matters: How does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130442. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef]
- Ruzzenente, B.; Rotig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, 115–122. [Google Scholar] [CrossRef]
- Spradling, A.; Ganetsky, B.; Hieter, P.; Johnston, M.; Olson, M.; Orr-Weaver, T.; Rossant, J.; Sanchez, A.; Waterston, R. New roles for model genetic organisms in understanding and treating human disease: Report from the 2006 Genetics Society of America meeting. Genetics 2006, 172, 2025–2032. [Google Scholar] [PubMed]
- Kabashi, E.; Brustein, E.; Champagne, N.; Drapeau, P. Zebrafish models for the functional genomics of neurogenetic disorders. Biochim. Biophys. Acta. 2011, 1812, 335–345. [Google Scholar] [CrossRef]
- Steele, S.L.; Prykhozhij, S.V.; Berman, J.N. Zebrafish as a model system for mitochondrial biology and diseases. Transl. Res. 2014, 163, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Wager, K.; Mahmood, F.; Russell, C. Modelling inborn errors of metabolism in zebrafish. J. Inherit. Metab. Dis. 2014, 37, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Postlethwait, J.; Ruotti, V.; Carvan, M.J.; Tonellato, P.J. Automated analysis of conserved syntenies for the zebrafish genome. Methods Cell Biol. 2004, 77, 255–271. [Google Scholar] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Gerety, S.S.; Wilkinson, D.G. Morpholino artifacts provide pitfalls and reveal a novel role for pro-apoptotic genes in hindbrain boundary development. Dev. Biol. 2011, 350, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Noyes, M.B.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 695–701. [Google Scholar] [CrossRef]
- Zu, Y.; Tong, X.; Wang, Z.; Liu, D.; Pan, R.; Li, Z.; Hu, Y.; Luo, Z.; Huang, P.; Wu, Q.; et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods 2013, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Burgess, S.M. Mutagenesis and phenotyping resources in zebrafish for studying development and human disease. Brief Funct. Genom. 2014, 13, 82–94. [Google Scholar] [CrossRef]
- Rafferty, S.A.; Quinn, T.A. A beginner’s guide to understanding and implementing the genetic modification of zebrafish. Prog. Biophys. Mol. Biol. 2018, 138, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Chlystun, M.; Campanella, M.; Law, A.L.; Duchen, M.R.; Fatimathas, L.; Levine, T.P.; Gerke, V.; Moss, S.E. Regulation of mitochondrial morphogenesis by annexin A6. PLoS ONE 2013, 8, e53774. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.Y.; Horng, J.L.; Kunkel, J.G.; Hwang, P.P. Proton pump-rich cell secretes acid in skin of zebrafish larvae. Am. J. Physiol. Cell Physiol. 2006, 290, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Wada, Y.; Kawamura, S. ES1 is a mitochondrial enlarging factor contributing to form mega-mitochondria in zebrafish cones. Sci. Rep. 2016, 6, 22360. [Google Scholar] [CrossRef] [PubMed]
- Drerup, C.M.; Herbert, A.L.; Monk, K.R.; Nechiporuk, A.V. Regulation of mitochondria-dynactin interaction and mitochondrial retrograde transport in axons. Elife 2017, 6, e22234. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.A.; Naylor, S.G.; Mercurio, S.; Dominguez, C.; Talbot, W.S. KBP is essential for axonal structure, outgrowth and maintenance in zebrafish, providing insight into the cellular basis of Goldberg-Shprintzen syndrome. Development 2008, 135, 599–608. [Google Scholar] [CrossRef]
- Campbell, P.D.; Shen, K.; Sapio, M.R.; Glenn, T.D.; Talbot, W.S.; Marlow, F.L. Unique function of Kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 2014, 34, 14717–14732. [Google Scholar] [CrossRef]
- Halpern, M.E.; Rhee, J.; Goll, M.G.; Akitake, C.M.; Parsons, M.; Leach, S.D. Gal4/UAS transgenic tools and their application to zebrafish. Zebrafish 2008, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.C.; Vargas, M.E.; Sagasti, A. WldS and PGC-1alpha regulate mitochondrial transport and oxidation state after axonal injury. J. Neurosci. 2013, 33, 14778–14790. [Google Scholar] [CrossRef]
- Plucinska, G.; Paquet, D.; Hruscha, A.; Godinho, L.; Haass, C.; Schmid, B.; Misgeld, T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 2012, 32, 16203–16212. [Google Scholar] [CrossRef]
- Auer, T.O.; Xiao, T.; Bercier, V.; Gebhardt, C.; Duroure, K.; Concordet, J.P.; Wyart, C.; Suster, M.; Kawakami, K.; Wittbrodt, J.; et al. Deletion of a kinesin I motor unmasks a mechanism of homeostatic branching control by neurotrophin-3. Elife 2015, 4, e05061. [Google Scholar] [CrossRef] [PubMed]
- Bergamin, G.; Cieri, D.; Vazza, G.; Argenton, F.; Mostacciuolo, M.L. Zebrafish Tg(hb9:MTS-Kaede): A new in vivo tool for studying the axonal movement of mitochondria. Biochim. Biophys. Acta. 2016, 1860, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Dukes, A.A.; Bai, Q.; van Laar, V.S.; Zhou, Y.Z.; Ilin, V.; David, C.N.; Agim, Z.S.; Bonkowsky, J.L.; Cannon, J.R.; Watkins, S.C.; et al. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP+ exposure. Neurobiol. of Disease 2016, 95, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Pinter, K.; Drerup, C.M. Analyzing Neuronal Mitochondria in vivo Using Fluorescent Reporters in Zebrafish. Front. Cell Dev. Biol. 2018, 6, 144. [Google Scholar] [CrossRef]
- Song, Y.; Selak, M.A.; Watson, C.T.; Coutts, C.; Scherer, P.C.; Panzer, J.A.; Gibbs, S.; Scott, M.O.; Willer, G.; Gregg, R.G.; et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD). PLoS ONE 2009, 4, e8329. [Google Scholar] [CrossRef]
- Dowling, J.J.; Arbogast, S.; Hur, J.; Nelson, D.D.; McEvoy, A.; Waugh, T.; Marty, I.; Lunardi, J.; Brooks, S.V.; Kuwada, J.Y.; et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 2012, 135, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Yang, X.; Ning, P.; Xi, X.; Yu, H.; Feng, Y.; Shao, R.; Meng, X. A mitochondria-targeted ratiometric two-photon fluorescent probe for detecting intracellular cysteine and homocysteine. Talanta 2018, 178, 24–30. [Google Scholar] [CrossRef]
- Bourdineaud, J.P.; Rossignol, R.; Brethes, D. Zebrafish: A model animal for analyzing the impact of environmental pollutants on muscle and brain mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2013, 45, 16–22. [Google Scholar] [CrossRef]
- Stackley, K.D.; Beeson, C.C.; Rahn, J.J.; Chan, S.S. Bioenergetic profiling of zebrafish embryonic development. PLoS ONE 2011, 6, e25652. [Google Scholar] [CrossRef]
- Artuso, L.; Romano, A.; Verri, T.; Domenichini, A.; Argenton, F.; Santorelli, F.M.; Petruzzella, V. Mitochondrial DNA metabolism in early development of zebrafish (Danio rerio). Biochim. Biophys. Acta. 2012, 1817, 1002–1011. [Google Scholar] [CrossRef]
- Lowery, L.A.; de Rienzo, G.; Gutzman, J.H.; Sive, H. Characterization and classification of zebrafish brain morphology mutants. Anat. Rec. 2009, 292, 94–106. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, K.H.; Kim, C.H.; Choi, S.Y. Real-time imaging of mitochondria in transgenic zebrafish expressing mitochondrially targeted GFP. Biotechniques 2008, 45, 331–334. [Google Scholar]
- Rendón, O.Z.; Neiva, L.S.; Sasarman, F.; Shoubridge, E.A. The arginine methyltransferase NDUFAF7 is essential for complex I assembly and early vertebrate embryogenesis. Hum. Mol. Genet. 2014, 23, 5159–5170. [Google Scholar] [CrossRef]
- Baden, K.N.; Murray, J.; Capaldi, R.A.; Guillemin, K. Early developmental pathology due to cytochrome c oxidase deficiency is revealed by a new zebrafish model. J. Biol. Chem. 2007, 282, 34839–34849. [Google Scholar] [CrossRef]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.; Gohil, V.M. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef]
- Kim, S.H.; Scott, S.A.; Bennett, M.J.; Carson, R.P.; Fessel, J.; Brown, H.A.; Ess, K.C. Multi-organ abnormalities and mTORC1 activation in zebrafish model of multiple acyl-CoA dehydrogenase deficiency. PLoS Genet. 2013, 9, e1003563. [Google Scholar] [CrossRef]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a Zebrafish Model of Dravet Syndrome. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Koster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef]
- Sallinen, V.; Kolehmainen, J.; Priyadarshini, M.; Toleikyte, G.; Chen, Y.C.; Panula, P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiol. Dis. 2010, 40, 93–101. [Google Scholar] [CrossRef]
- Flinn, L.J.; Keatinge, M.; Bretaud, S.; Mortiboys, H.; Matsui, H.; de Felice, E.; Woodroof, H.I.; Brown, L.; McTighe, A.; Soellner, R.; et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol. 2013, 74, 837–847. [Google Scholar] [CrossRef]
- Noble, S.; Ismail, A.; Godoy, R.; Xi, Y.; Ekker, M. Zebrafish Parla- and Parlb-deficiency affects dopaminergic neuron patterning and embryonic survival. J. Neurochem. 2012, 122, 196–207. [Google Scholar] [CrossRef]
- Bretaud, S.; Allen, C.; Ingham, P.W.; Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 2007, 100, 1626–1635. [Google Scholar] [CrossRef]
- Froyset, A.K.; Edson, A.J.; Gharbi, N.; Khan, E.A.; Dondorp, D.; Bai, Q.; Tiraboschi, E.; Suster, M.L.; Connolly, J.B.; Burton, E.A.; et al. Astroglial DJ-1 over-expression up-regulates proteins involved in redox regulation and is neuroprotective in vivo. Redox. Biol. 2018, 16, 237–247. [Google Scholar] [CrossRef]
- Sheng, D.; Qu, D.; Kwok, K.H.; Ng, S.S.; Lim, A.Y.; Aw, S.S.; Lee, C.W.; Sung, W.K.; Tan, E.K.; Lufkin, T.; et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 2010, 6, e1000914. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.C.; Lulla, A.; Stahl, M.C.; Wheat, N.D.; Bronstein, J.M.; Sagasti, A. Axon degeneration and PGC-1alpha-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis. Model Mech. 2014, 7, 571–582. [Google Scholar] [CrossRef]
- Milanese, C.; Sager, J.J.; Bai, Q.; Farrell, T.C.; Cannon, J.R.; Greenamyre, J.T.; Burton, E.A. Hypokinesia and reduced dopamine levels in zebrafish lacking beta- and gamma1-synucleins. J. Biol. Chem. 2012, 287, 2971–2983. [Google Scholar] [CrossRef]
- Vettori, A.; Bergamin, G.; Moro, E.; Vazza, G.; Polo, G.; Tiso, N.; Argenton, F.; Mostacciuolo, M.L. Developmental defects and neuromuscular alterations due to mitofusin 2 gene (MFN2) silencing in zebrafish: A new model for Charcot-Marie-Tooth type 2A neuropathy. Neuromuscul. Disord. 2011, 21, 58–67. [Google Scholar] [CrossRef]
- Chapman, A.L.; Bennett, E.J.; Ramesh, T.M.; de Vos, K.J.; Grierson, A.J. Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS ONE 2013, 8, e67276. [Google Scholar] [CrossRef]
- Rahn, J.J.; Stackley, K.D.; Chan, S.S. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE 2013, 8, e59218. [Google Scholar] [CrossRef]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; de Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1 are Associated with Impaired Neuromuscular Transmission. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Steffen, J.; Yourshaw, M.; Mamsa, H.; Andersen, E.; Rudnik-Schoneborn, S.; Pope, K.; Howell, K.B.; McLean, C.A.; Kornberg, A.J.; et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain 2016, 139, 2877–2890. [Google Scholar] [CrossRef]
- Pei, W.; Kratz, L.E.; Bernardini, I.; Sood, R.; Yokogawa, T.; Dorward, H.; Ciccone, C.; Kelley, R.I.; Anikster, Y.; Burgess, H.A.; et al. A model of Costeff Syndrome reveals metabolic and protective functions of mitochondrial OPA3. Development 2010, 137, 2587–2596. [Google Scholar] [CrossRef][Green Version]
- Taylor, M.R.; Hurley, J.B.; van Epps, H.A.; Brockerhoff, S.E. A zebrafish model for pyruvate dehydrogenase deficiency: Rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc. Natl. Acad. Sci. USA 2004, 101, 4584–4589. [Google Scholar] [CrossRef]
- Kasher, P.R.; Namavar, Y.; van Tijn, P.; Fluiter, K.; Sizarov, A.; Kamermans, M.; Grierson, A.J.; Zivkovic, D.; Baas, F. Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum. Mol. Genet. 2011, 20, 1574–1584. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Koopman, W.J.; Distelmaier, F.; Smeitink, J.A.; Willems, P.H. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta. 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Chen, S.; Wu, Y.; Lin, S.; Duan, Y.; Zheng, K.; Zhang, L.; Gu, X.; Hong, W.; et al. A Novel Potentially Causative Variant of NDUFAF7 Revealed by Mutation Screening in a Chinese Family With Pathologic Myopia. Invest. Ophthalmol. Vis. Sci. 2017, 58, 4182–4192. [Google Scholar] [CrossRef]
- DiMauro, S.; Tanji, K.; Schon, E.A. The many clinical faces of cytochrome c oxidase deficiency. Adv. Exp. Med. Biol. 2012, 748, 341–357. [Google Scholar]
- Olsen, R.K.; Andresen, B.S.; Christensen, E.; Bross, P.; Skovby, F.; Gregersen, N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003, 22, 12–23. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Jones, N. Mitochondrial dysfunction occurs early in PD. Nat. Rev. Neurol. 2010, 6, 60. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Xi, Y.; Noble, S.; Ekker, M. Modeling neurodegeneration in zebrafish. Curr. Neurol. Neurosci. Rep. 2011, 11, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Tuimala, J.; Chen, Y.C.; Panula, P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol. Dis. 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Soman, S.; Keatinge, M.; Moein, M.; da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(−/−) zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, D.T.; Olzomer, E.M.; Poon, G.P.; Cole, N.J.; Puvanendran, A.; Phillips, B.R.; Hesselson, D. Rescue of Pink1 Deficiency by Stress-Dependent Activation of Autophagy. Cell Chem. Biol. 2017, 24, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Lee, J.R.; Grimes, D.A.; Racacho, L.; Ye, D.; Yang, H.; Ross, O.A.; Farrer, M.; McQuibban, G.A.; Bulman, D.E. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 1966–1974. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Rogozin, I.B.; Davidovic, L.; Letellier, M.C.; Pellegrini, L. The rhomboids: A nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003, 4, 19. [Google Scholar] [CrossRef]
- Bai, Q.; Mullett, S.J.; Garver, J.A.; Hinkle, D.A.; Burton, E.A. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006, 1113, 33–44. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [PubMed]
- Angeles, D.C.; Gan, B.H.; Onstead, L.; Zhao, Y.; Lim, K.L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K. alpha-Synuclein and mitochondria: Partners in crime? Neurotherapeutics 2013, 10, 391–399. [Google Scholar] [CrossRef]
- Sun, Z.; Gitler, A.D. Discovery and characterization of three novel synuclein genes in zebrafish. Dev. Dyn. 2008, 237, 2490–2495. [Google Scholar] [CrossRef] [PubMed]
- Lovas, J.R.; Wang, X. The meaning of mitochondrial movement to a neuron’s life. Biochim. Biophys. Acta. 2013, 1833, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.; Brown, R.; Dobyns, W.B.; Harington, J.; Patel, J.; Quinn, M.; Brown, G. Pontocerebellar hypoplasia type 6: A British case with PEHO-like features. Am. J. Med. Genet. A. 2010, 152, 2079–2084. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.D.; Marlow, F.L. Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene. Expr. Patterns. 2013, 13, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.S.; Mayor, J.A.; Wood, D.O. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J. Biol. Chem. 1993, 268, 13682–13690. [Google Scholar] [PubMed]
- Moraes, T.F.; Reithmeier, R.A. Membrane transport metabolons. Biochim. Biophys. Acta. 2012, 1818, 2687–2706. [Google Scholar] [CrossRef] [PubMed]
- Morciano, P.; Carrisi, C.; Capobianco, L.; Mannini, L.; Burgio, G.; Cestra, G.; de Benedetto, G.E.; Corona, D.F.; Musio, A.; Cenci, G. A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum. Mol. Genet. 2009, 18, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef]
- Powell, K.A.; Davies, J.R.; Taylor, E.; Wride, M.A.; Votruba, M. Mitochondrial localization and ocular expression of mutant Opa3 in a mouse model of 3-methylglutaconicaciduria type III. Invest. Ophthalmol. Vis. Sci. 2011, 52, 4369–4380. [Google Scholar] [CrossRef]
- Patel, M.S.; Roche, T.E. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990, 4, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, T.; Baas, F.; Barth, P.G.; Poll-The, B.T. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet. J. Rare Dis. 2018, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Grundlingh, J.; Dargan, P.I.; El-Zanfaly, M.; Wood, D.M. 2,4-dinitrophenol (DNP): A weight loss agent with significant acute toxicity and risk of death. J. Med. Toxicol. 2011, 7, 205–212. [Google Scholar] [CrossRef]
- Bestman, J.E.; Stackley, K.D.; Rahn, J.J.; Williamson, T.J.; Chan, S.S. The cellular and molecular progression of mitochondrial dysfunction induced by 2,4-dinitrophenol in developing zebrafish embryos. Differentiation 2015, 89, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166. [Google Scholar] [CrossRef]
- Ferriero, R.; Manco, G.; Lamantea, E.; Nusco, E.; Ferrante, M.I.; Sordino, P.; Stacpoole, P.W.; Lee, B.; Zeviani, M.; Brunetti-Pierri, N. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci. Transl. Med. 2013, 5, 175ra31. [Google Scholar] [CrossRef]
- Maurer, C.M.; Schonthaler, H.B.; Mueller, K.P.; Neuhauss, S.C. Distinct retinal deficits in a zebrafish pyruvate dehydrogenase-deficient mutant. J. Neurosci. 2010, 30, 11962–11972. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Schiavone, M.; Galber, C.; Carini, M.; da Ros, T.; Petronilli, V.; Argenton, F.; Carelli, V.; Acosta Lopez, M.J.; Salviati, L.; et al. The idebenone metabolite QS10 restores electron transfer in complex I and coenzyme Q defects. Biochim. Biophys. Acta. Bioenerg. 2018, 1859, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Leber’s hereditary optic neuropathy misdiagnosed as optic neuritis and Lyme disease in a patient with multiple sclerosis. BMJ Case Rep. 2018, 11, e227109. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Geng, W.S.; Jia, J.J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef]
- Wen, L.; Wei, W.; Gu, W.; Huang, P.; Ren, X.; Zhang, Z.; Zhu, Z.; Lin, S.; Zhang, B. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev. Biol. 2008, 314, 84–92. [Google Scholar] [CrossRef]
- Lu, X.L.; Lin, Y.H.; Wu, Q.; Su, F.J.; Ye, C.H.; Shi, L.; He, B.X.; Huang, F.W.; Pei, Z.; Yao, X.L. Paeonolum protects against MPP+-induced neurotoxicity in zebrafish and PC12 cells. BMC Complement. Altern. Med. 2015, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Casado, M.E.; Lima, E.; Garcia, J.A.; Doerrier, C.; Aranda, P.; Sayed, R.K.; Guerra-Librero, A.; Escames, G.; Lopez, L.C.; Acuna-Castroviejo, D. Melatonin rescues zebrafish embryos from the parkinsonian phenotype restoring the parkin/PINK1/DJ-1/MUL1 network. J. Pineal. Res. 2016, 61, 96–107. [Google Scholar] [CrossRef]
- Srinivasan, V.; Cardinali, D.P.; Srinivasan, U.S.; Kaur, C.; Brown, G.M.; Spence, D.W.; Hardeland, R.; Pandi-Perumal, S.R. Therapeutic potential of melatonin and its analogs in Parkinson’s disease: Focus on sleep and neuroprotection. Ther. Adv. Neurol. Disord. 2011, 4, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Loddo, G.; Calandra-Buonaura, G.; Sambati, L.; Giannini, G.; Cecere, A.; Cortelli, P.; Provini, F. The Treatment of Sleep Disorders in Parkinson’s Disease: From Research to Clinical Practice. Front. Neurol. 2017, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.W.; Hung, H.C.; Huang, S.Y.; Chen, C.H.; Chen, Y.R.; Chen, C.Y.; Yang, S.N.; Wang, H.D.; Sung, P.J.; Sheu, J.H.; et al. Neuroprotective Effect of the Marine-Derived Compound 11-Dehydrosinulariolide through DJ-1-Related Pathway in In Vitro and In Vivo Models of Parkinson’s Disease. Mar. Drugs 2016, 14, 187. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Z.; Cui, W. Marine-Derived Natural Compounds for the Treatment of Parkinson’s Disease. Mar. Drugs 2019, 17, 221. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Tools | Investigation | Refs |
|---|---|---|
| MitoTracker Deep Red and MitoTracker Green FM | Mitochondrial activity in skin | [34] |
| mitochondrial localization sequence- enhanced GFP embryos | Mitochondrial morphology in real time | [53] |
| Mito:mCherry | KBP/KIAA1279 function | [37] |
| Oxidative fluorescent dye dihydrorodamine-123 (DHR-123) | Mitochondrial ROS formation | [46] |
| Seahorse XF24 Extracellular Flux Analyzer | Mitochondrial bioenergetics | [50] |
| Relative quantification by RT-PCR and immuno-probes | Genes involved in mtDNA replication and transcription and genes of the OxPhos system | [51] |
| MitoSOXTM | Examination of mitochondria-derived ROS | [47] |
| MitoFish, Tg(elavl3.2:Gal4-VP16)mde4/Tg(UAS-E1b:mYFP,mitoCFP)mde | Time-lapse imaging of mitochondrial transport in Rohon-Beard sensory neurons | [41] |
| MitoDsRed transgenic line: reporter UAS–mitoDsRed–polyA crossed to isl1(ss):Gal4VP16:14XUAS-GFP fish. | Morphology and motility of mitochondria in somatosensory neurons | [40] |
| MitoGFP | In vivo study of mitochondria in retinal ganglion cell of kif5Aa mutant | [42] |
| Tg(hb9:MTS-Kaede) | Mitochondrial dynamics in motor neurons in CMT2A mutants | [43] |
| Tg(otpb:Gal4); Tg(UAS:mtPAGFP:mtDsRed2) | Measurement of mitochondrial transport in dopaminergic neurons | [44] |
| Anti-mitochondrial membrane 20 (TOM20) and anti-mitochondrial aspartate aminotransferase (mAAT)antibodies | Study of ES1, a mitochondria-enlarging factor in cones | [35] |
| 5kbneurod:mito-TagRFP plasmid | Role for Actr10 in dynactin-mitochondria interaction | [36] |
| Tg(5kbneurod:mitomEos)y586 | Mitochondrial lifetime and the timeline of mitochondrial turnover and temporal dynamics of mitochondrial gain and loss from axon terminals | [45] |
| 5kbneurod:mito-Timer | ROS production, an indicator of mitochondrial health | [45] |
| Vital dye TMRE | Measurement of potential/pH across the mitochondrial inner membrane | [45] |
| G-GECO1; Tg(5kbneurod:GGECO)nl19; R-GECO1; Tg(5kbneurod:mito-R-GECO)nl20 | Mitochondrial calcium buffering in neurons | [45] |
| ATP:ADP dual ratiometric sensor PercevalHR | Productivity of mitochondria in the cell body and axon terminal of pLL sensory axons | [45] |
| Mito-MQ | Oxidative stress by Real-time monitoring Cys and Hcy levels | [48] |
| Gene | Function | Modeling Zebrafish | Phenotype | Refs |
|---|---|---|---|---|
| Defects of functioning and assembly of OxPhos complex | ||||
| NDUFAF7 | Assembly factor of Complex I | MO-mediated knockdown | Delayed hatching times and morphological abnormality | [54] |
| COXVa | Structural component of COX | MO-mediated knockdown | Developmental defects in endodermal tissue, cardiac function and swimming behavior | [55] |
| SURF1 | Assembly factor of COX | MO-mediated knockdown | Developmental defects in endodermal tissue, cardiac function and swimming behavior | [55] |
| COA6 | Cu-delivery pathway for COX assembly | MO-mediated knockdown | Heart developmental defects | [56] |
| ETFDH | Electron transfer flavoprotein dehydrogenase | xavier mutant zebrafish line | Altered energy metabolism, dysregulated ROS production, increased aerobic glycolysis, motility defects, abnormal glial patterning, reduced motor axon branching and neuromuscular synapse number | [46] |
| MO-mediated knockdown | Bent tail and reduced heartbeat, aberrant swimming behavior, and reduced neuromuscular synaptogenesis | [46] | ||
| ETFA | Receiving of electrons from dehydrogenases involved in fatty acid β-oxidation, amino acid and choline metabolism | dark xavier (dxavu463) mutant zebrafish line | Increased number of neural progenitor cells and accumulation of neutral lipid and cerebroside sulphate in brain, hepatic steatosis and dysmorphic kidneys, and hypomyelination | [57] |
| Ion channels and mitochondrial defects | ||||
| SCN1A | Brain-specific voltage-activated sodium channel | scn1Lab mutant zebrafish line | Increased behavioral seizure activity and increased glycolytic rate | [58] |
| Defects of mitochondrial quality control system | ||||
| PARK2 | E3 ubiquitin ligase | MO-mediated knockdown | High susceptibility to the MPP+, Dopaminergic loss neurons and complex I deficiency | [59] |
| Overexpression of parkin in transgenic zebrafish cell lines | Protective reaction against cell death induced by proteotoxic stress | [60] | ||
| PINK1 | Mitochondrial serine/threonine kinase | MO-mediated knockdown | High sensitivity to MPTP, increase of ROS level, activation of apoptotic signaling | [61] |
| pink1 null mutant zebrafish line | PD-phenotype and altered biogenesis of mitochondria | [62] | ||
| PARL | Presenilin-associated rhomboid-like protein that is part of the PINK1 and Parkin pathway. In zebrafish are present two paralogs (parla and parlb) | MO-mediated knockdown | High rate of mortality in early larvae, mis-patterned dopaminergic neurons in morphants | [63] |
| PARK7 | Pleiotropic function: transcriptional regulator, antioxidant scavenger, redox sensor and roles as a chaperone with protease activity | MO-mediated knockdown | Neurons are highly sensitive to hydrogen peroxidase | [64] |
| Transgenic line Tg(gfap:egfp-2A-flag-zDJ-1) | Astrocytic dj-1 overexpression protects mutants from neurological damage induced by the PD-related neurotoxin MPP+ | [65] | ||
| LRRK2 | Cytosolic protein | MO-mediated knockdown | Neurodegeneration and locomotion defects | [66] |
| SNCA | Soluble protein primarily expressed in the neural tissue. In zebrafish are present only two synuclein isoforms, β and γ2-synuclein | Overexpression of human α-synuclein | Changes in mitochondrial density and morphology, mitochondrial fragmentation, reduced mitochondrial motility and ensuing synaptic dysfunction and degeneration | [67] |
| Double MO-mediated knockdown | Motor impairments and reduction of dopamine level | [68] | ||
| Defects of mitochondrial dynamics | ||||
| MFN2 | Promotion of mitochondria movement and fusion | MO-mediated knockdown | Reduction of the survival rate, motor impairment or unresponsiveness to touch. Generalized impairment in the axonal structure of primary motor neurons. Presence of shortened or missing axons, altered distribution of acetylcholine receptors. Altered alignment of the myofibers | [69] |
| mfn2L285X mutant zebrafish line | Altered swimming and progressive loss of motor function. Alterations at the neuromuscular junction. Altered mitochondrial dynamics and change in mitochondrial morphology | [70] | ||
| Overexpression of MFN2 carrying the c.281G>A (R94Q) and c.227T>C (L76P) mutations in transgenic zebrafish cell lines | Reduction of mitochondria transport along the axon in p.R94Q expressing larvae. Reduction of density of moving mitochondria in the case of p.L76P overexpression. | [43] | ||
| OPA1 | Fusion of the outer and inner mitochondrial membranes | MO-mediated knockdown | Developmental delay, decreasing of the blood circulation velocity, and reduction of the eye size and the heart rate. Reduction of the startle response and bioenergetics defects | [71] |
| KBP/KIAA1279 | Binding protein of the kinesin motor proteins KIF1B and KIF1C | kbpst23 mutants | Delay in development of peripheral axons. Axons degeneration. Reduction in myelination. Disorganization of the axonal cytoskeleton. Reduction in the number of axonal mitochondria. | [37] |
| MO-mediated knockdown | Axonal defects in peripheral and central nervous systems | [37] | ||
| KIF5A | Transport processes | kif5Aa mutant zebrafish line | Hyperexcitability, peripheral polyneuropathy, and axonal degeneration | [38] |
| Mitochondrial carrier deficiency | ||||
| SLC25A1 | Mitochondrial solute carrier family | MO-mediated knockdown | Altered tail morphology, impaired swimming and touch-evoked escape responses. Abnormal neuromuscular junction development. Short and erratic outgrowth toward the muscle fiber of the motor axon terminals with no complete synapse formation. Hindbrain, heart, yolk sac, and tail edema. Abnormal heart development with reduced blood flow to the tail (in severe phenotypes) | [72] |
| SLC25A46 | Mitochondrial solute carrier family | MO-mediated knockdown | Curly-tail morphology and altered swimming. Shorter axon tracts in motor neurons and fewer neuronal processes in spinal cord. Degenerate neurons with incomplete mitochondrial fission, mitochondria aggregated and misplaced. | [73] |
| MO-mediated knockdown | Poor motility and the loss of spinal motor neurons. | [74] | ||
| Metabolic mitochondrial disorders | ||||
| OPA3 | Inner mitochondrial membrane protein | MO-mediated Knockdown | Increased 3-methylglutaconic acid (MGC), and features mimicking movement disorders | [75] |
| PDHC | Nuclear-encoded mitochondrial matrix multienzyme complex | Spontaneous zebrafish mutant (noa strain, carrying a mutation in the pdh-e2 subunit) | Retinal abnormalities, defects of synaptic transmission and of light adaptation in cone photoreceptor cells, premature death, lethargy, expanded melanophores, and absence of feeding behavior | [76] |
| TSEN54 | Subunit of the tRNA-splicing endonuclease (TSEN) complex | MO-mediated Knockdown, mutagenesis strategy | Brain hypoplasia and loss of structural integrity, increased cell death, and early lethality in zebrafish | [77] |
| RARS2 | Mitochondrial arginyl-tRNA synthetase gene | MO-mediated knockdown | Brain hypoplasia, cell death and neurodegeneration | [77] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fichi, G.; Naef, V.; Barca, A.; Longo, G.; Fronte, B.; Verri, T.; Santorelli, F.M.; Marchese, M.; Petruzzella, V. Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System. Int. J. Mol. Sci. 2019, 20, 2409. https://doi.org/10.3390/ijms20102409
Fichi G, Naef V, Barca A, Longo G, Fronte B, Verri T, Santorelli FM, Marchese M, Petruzzella V. Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System. International Journal of Molecular Sciences. 2019; 20(10):2409. https://doi.org/10.3390/ijms20102409
Chicago/Turabian StyleFichi, Gianluca, Valentina Naef, Amilcare Barca, Giovanna Longo, Baldassare Fronte, Tiziano Verri, Filippo M. Santorelli, Maria Marchese, and Vittoria Petruzzella. 2019. "Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System" International Journal of Molecular Sciences 20, no. 10: 2409. https://doi.org/10.3390/ijms20102409
APA StyleFichi, G., Naef, V., Barca, A., Longo, G., Fronte, B., Verri, T., Santorelli, F. M., Marchese, M., & Petruzzella, V. (2019). Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System. International Journal of Molecular Sciences, 20(10), 2409. https://doi.org/10.3390/ijms20102409