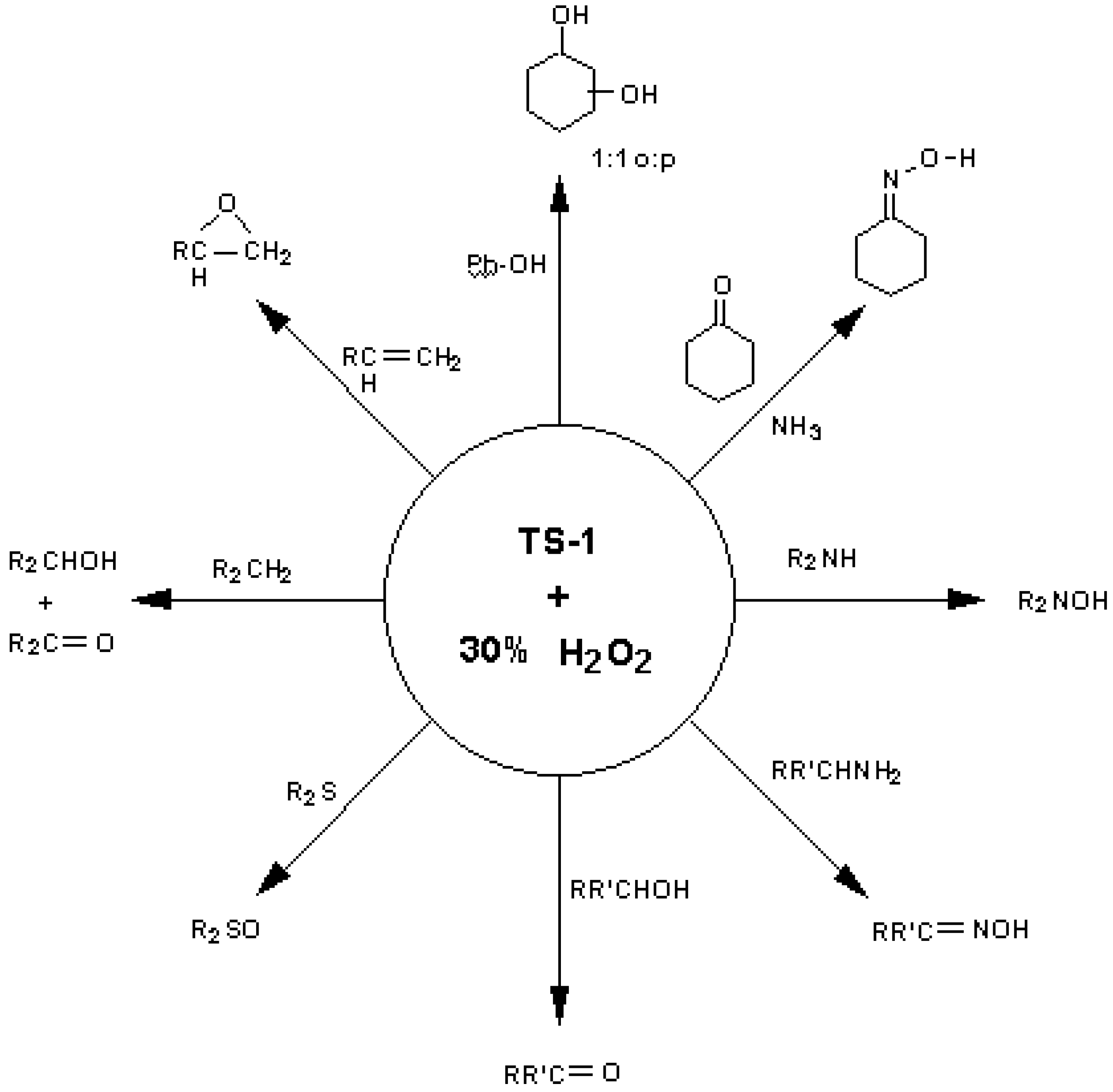
3. The TS-1 case study
TS-1 catalyst was proposed in the early eighty [
1] and since then many attempts have been made in order to understand the role of titanium centers in the catalytic process. In the eighties and in the begin of the nineties a lively debate has been observed in the literature about the structural nature of the Ti centers in TS-1: titanyl groups, extraframework defect sites, monomeric and dimeric Ti species, Ti species incorporated in edge sharing type structures forming bridges across the zeolite channels, have been inferred by different authors; the same holds for the local geometries, where Ti species having local coordinations like tetrahedral, square pyramidal, and octahedral have been hypothesized.
The origin of the initial confusion was probably related with the difficulty encountered in the synthesis of well manufactured TS-1's, which requires the use of extremely pure reagents and severe control in the synthesis conditions [
1]. An imperfect synthesis implies an incomplete incorporation of Ti into the MFI framework, leading to a significant reduction of the catalytic performances and possibly to a misinterpretation of structural and spectroscopic data. Moreover, the fact that only a very small amount of Ti (less than 3 wt. % in TiO
2, corresponding to a molar ratio of
x = [Ti]/([Ti]+[Si]) = 0.025 [
19]) can be substitutionally incorporated into the MFI framework [
20] does not facilitate the extraction and the attribution of the Ti contribution from the total experimental signal. Note that the
x = 0.025 limit has been recently slightly improved [
21,
22].
Nowadays there is a general consensus that the Ti(IV) atoms are incorporated as isolated centers into the framework and are substituting Si atoms in the tetrahedral positions, as schematized in
Figure 3a. The model of isomorphous substitution has been put forward on the basis of several independent characterization techniques, namely X-rays [
19,
23,
24] or neutron [
21,
25,
26] diffraction studies, IR (Raman) [
22,
27,
28,
29,
30,
31,
32], UV-Vis [
30,
33,
34], EXAFS and XANES [
21,
34,
35,
36,
37,
38,
39,
40] spectroscopies. Points
i)-
vii) will be devoted to a brief discussion of each technique.
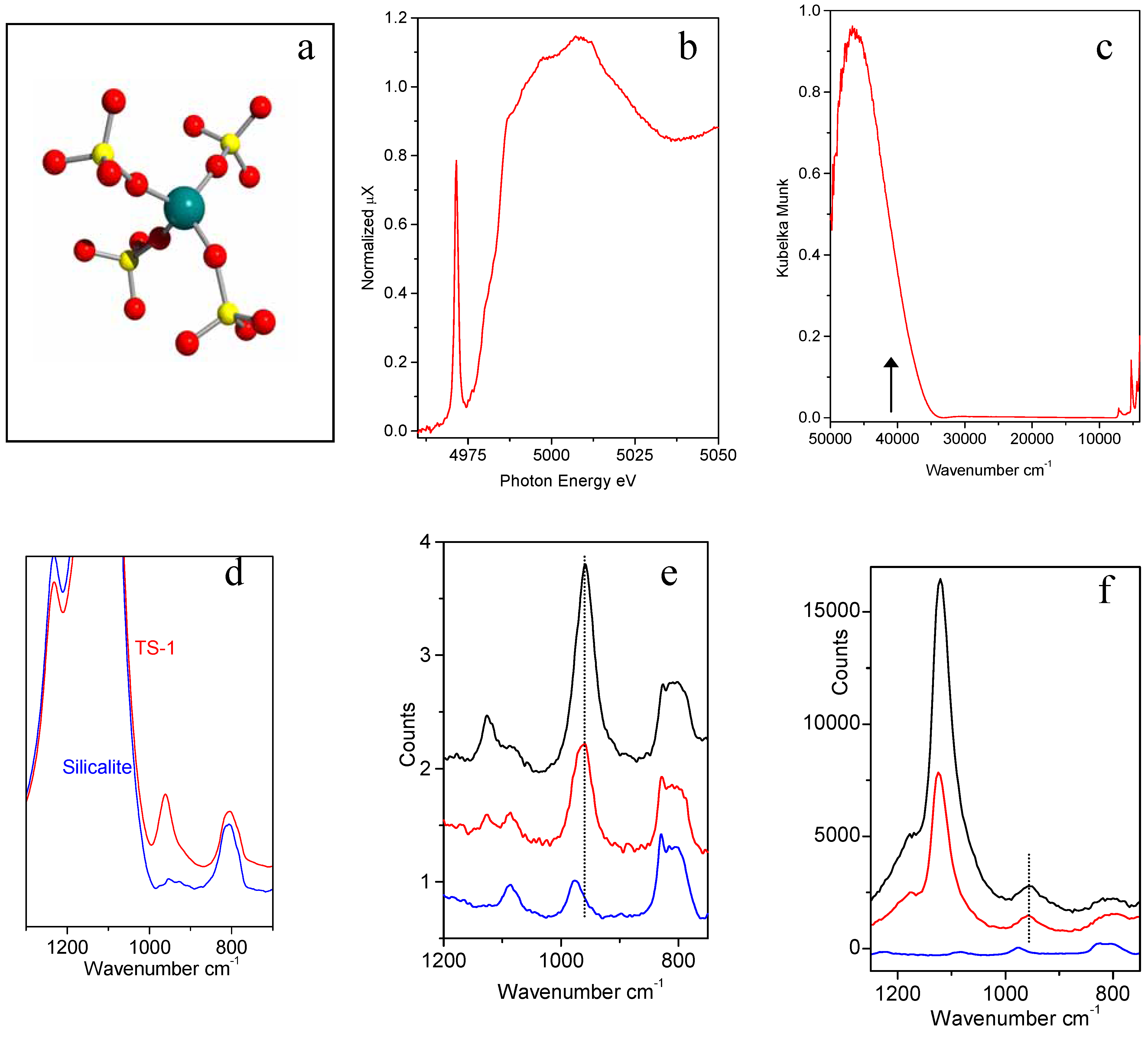
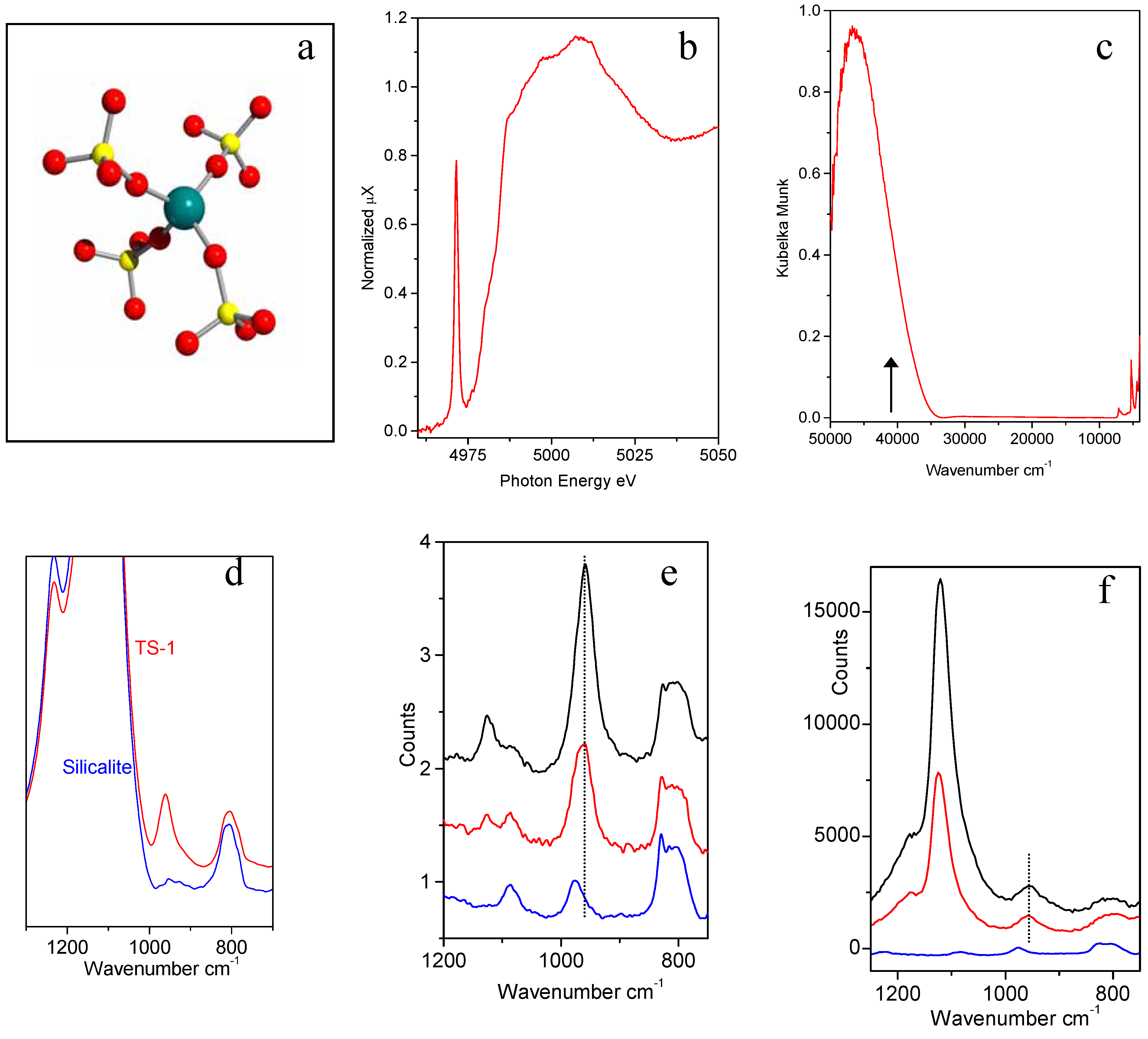
i) XANES spectroscopy shows that a narrow and intense pre-edge peak at 4967 eV due to the 1
s → 3
d electronic transition involving Ti atoms in tetrahedral coordination is present in well manufactured TS-1 (
Figure 3b). Conversely, the 1
s → 3
d electronic transition of Ti(IV) species in TiO
2 (anatase or rutile) is characterized by a very low intensity. Indeed the transitions A
1g → T
2g are symmetrically forbidden in the case of octahedral coordination of Ti (IV), but the transition A
1→ T
2 is allowed in the case of tetrahedral coordination of Ti (IV), as in the case of [TiO
4] units [
22,
34,
35,
36,
38,
40].
ii) The UV-vis spectra of TS-1
in vacuo gives a simple and clear proof of the presence of tetrahedral Ti(IV) in the zeolite framework [
22,
30,
32,
33,
34]: in fact a Ti
4+O
2-→ Ti
3+O
- ligand to metal charge transfer (LMCT) located at ≈48.000 cm
-1 (
Figure 3c) can be unambiguously assigned to the charge transfer transition from the oxygen ligand to an unoccupied orbital of a Ti(IV) ion tetrahedrally coordinated in isolated [TiO
4]. In fact Ti(IV) species in octahedral coordination (like in anatase or rutile) exhibit a Ti
4+O
2- → Ti
3+O
- LMCT in the 31500-33000 cm
-1;
iii) the IR spectra of TS-1 in the framework stretching region shows an absorption band located at 960 cm
-1 (red curve in
Figure 3d), virtually absent in perfect silicalite (blue curve in
Figure 3d) and immediately identified as a fingerprint of TS-1 [
22,
27,
28,
29,
30,
31]. A qualitative correlation between the intensity of the infrared band at 960 cm
-1 and titanium content has been observed since the first synthesis of TS-1. Indeed, the occurrence of that band is one of the distinctive features of the material cited in the original patent [
1]. However, the quantitative correlation has been reported only very recently [
22]. In the same work the nature of the 960 cm
-1 band has been discussed in terms of both cluster and periodical approaches.
iv) In the early nineties Raman spectroscopy has been applied to the characterization of TS-1 catalyst [
29,
31]. In such experiments, beside the 960 cm
-1 band, already observed by IR spectroscopy (see point
iii), a new component at 1125 cm
-1 has been detected. Also the 1125 cm
-1 band was recognized to be a fingerprint of the insertion of Ti atoms in the zeolitic framework [
29,
31].
v) Very recently, Li
et al. have reported an interesting resonant Raman study of TS-1 [
32]. They have used a λ = 244 nm (40984 cm
-1) laser source instead of the conventional near-IR laser at λ = 1064 nm (9398 cm
-1). The frequency of the UV laser is located on the low energy tail of the Ti
4+O
2- → Ti
3+O
- LMCT of Ti(IV) species in TS-1, see black arrow in
Figure 3c. This implies that a resonant Raman enhancement is expected for the vibrational modes related to Ti species. Li
et al. [
32] have reported that, under such conditions, only the 1125 cm
-1 component undergoes the expected enhancement, while the 960 cm
-1 one does not (
Figure 3f). The enhancement of the intensity of the 1125 cm
-1 feature and the invariance of the 960 cm
-1 feature in UV-Raman experiments, must be discussed in terms of resonant Raman selection rules [
22]. Quantum chemical calculations on cluster models. Si[OSi(OH)
3]
4 and Ti[OSi(OH)
3]
4 (
Figure 3a) at the B3LYP/6-31G(d) level of theory provide the basis for the assignment of the main vibrational contributions, and for the understanding of Raman enhancement. The resonance-enhanced 1125 cm
-1 mode is unambiguously associated with a totally symmetric vibration of the TiO
4 tetrahedron, achieved through in-phase antisymmetric stretching of the four connected Ti-O-Si bridges [
22]. This vibration can also be described as a totally symmetric stretching of the four Si-O bonds pointing towards Ti. The resonance enhancement of this feature is explained in terms of the electronic structure of the Ti-containing moiety. Asymmetric stretching modes of TO
4 units show distinct behavior when T is occupied by Si or Ti. Calculations on periodic models of silicalite and TS-1 free of OH groups using the QMPOT embedding method, correctly reproduce the transparency gap of silicalite and the appearance of asymmetric TiO
4 vibrations at 960 cm
-1 in TS-1 [
22]. This study has definitively proved that the 960 cm
-1 band could not be confused with the broader IR absorption observed around 975 cm
-1 in defective silicalites (
i.e. silicalites hosting internal hydroxylated nests caused by Si vacancies). The latter component can be appreciated in the blue spectra reported in parts e) and f) of
Figure 3 and have been extensively debated in refs. [
22,
41,
42,
43].
Figure 3.
Part (a): schematic representation of the insertion of Ti in the Ti in the zeolitic [Ti(OSiO3)4] unit. Ti, Si and O atoms are represented by blue, yellow and red spheres respectively. Parts (b-f): XANES, UV-Vis, IR Raman and resonant Raman features typical of [Ti(OSiO3)4] units. Blue, red ad black curves refers to Ti-free silicalite, and to TS-1 with 1.4 and 3.0 wt. % in TiO2, respectively.
Figure 3.
Part (a): schematic representation of the insertion of Ti in the Ti in the zeolitic [Ti(OSiO3)4] unit. Ti, Si and O atoms are represented by blue, yellow and red spheres respectively. Parts (b-f): XANES, UV-Vis, IR Raman and resonant Raman features typical of [Ti(OSiO3)4] units. Blue, red ad black curves refers to Ti-free silicalite, and to TS-1 with 1.4 and 3.0 wt. % in TiO2, respectively.
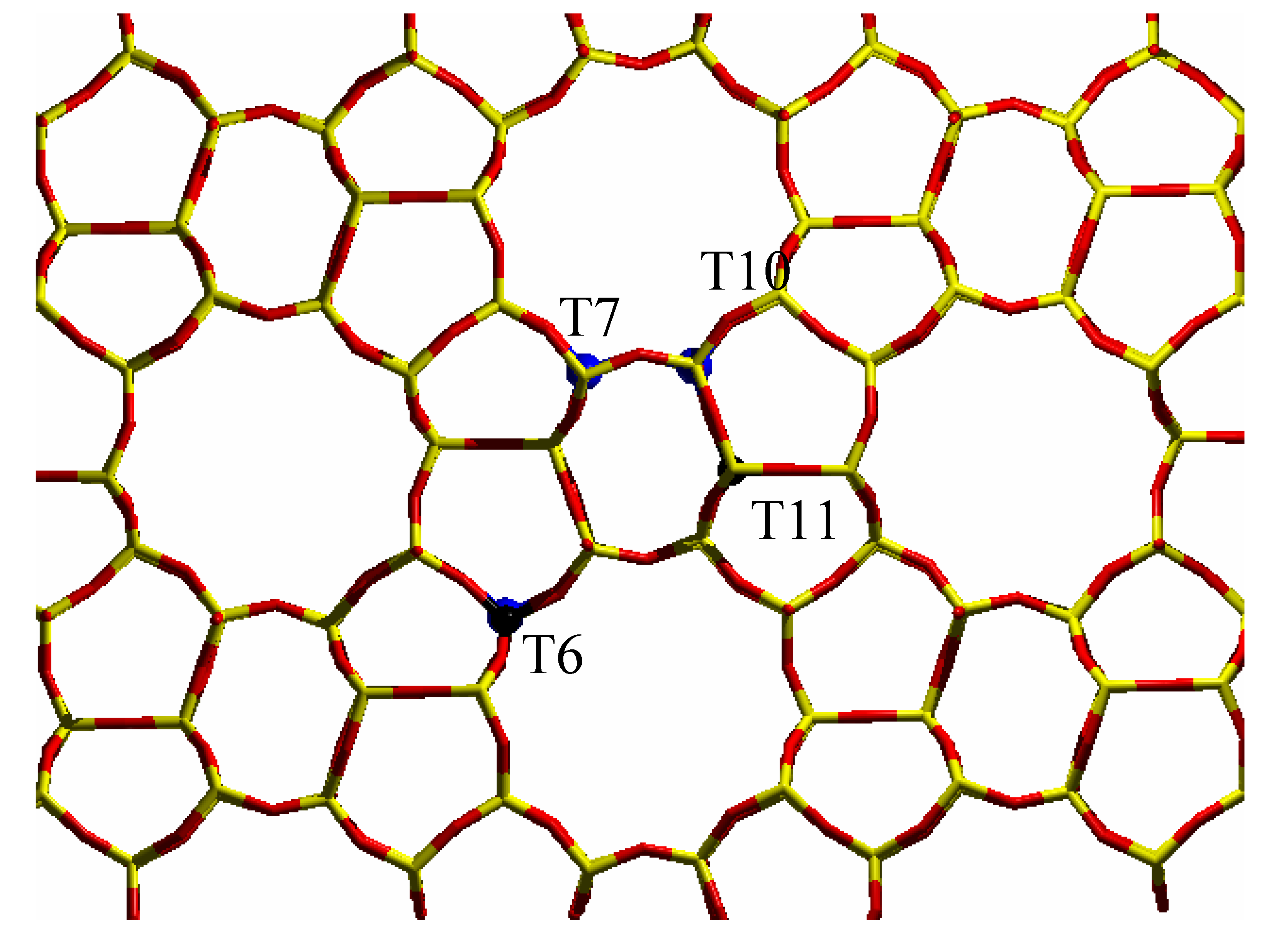
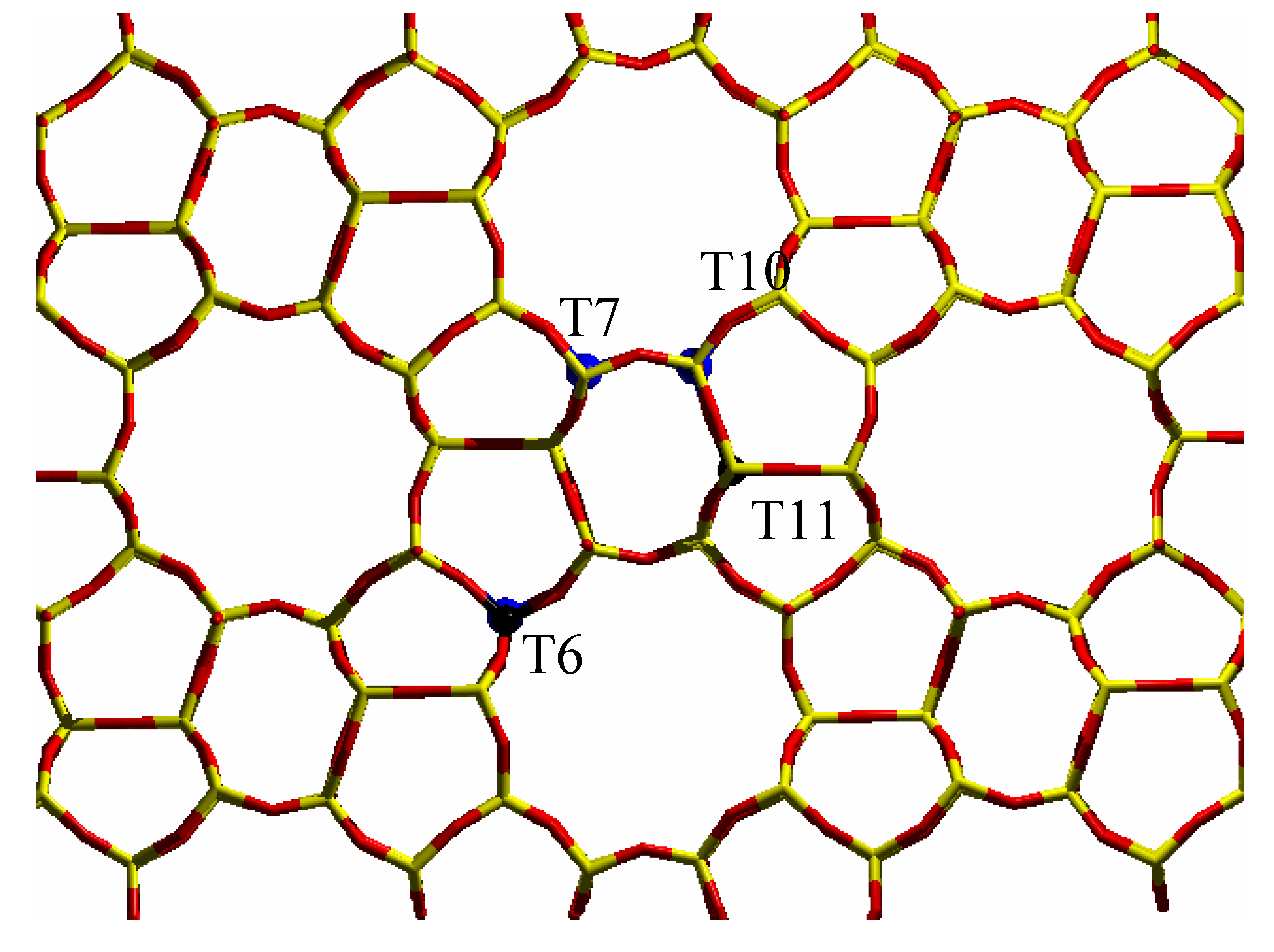
vi) diffraction data show that the unit cell volume of silicalite increases linearly with Ti content when Ti(IV) atoms are inserted in the framework [
19,
23]. Very recently, diffraction methods have been employed to try to discuss the problem of Ti partitioning among the 12 crystallographically unequivalent T sites [
21,
23,
24,
25,
26]. The aim of such studies was to determine if Ti atoms are equally shared among the 12 T sites, or if one ore more preferential substitution sites are present. Such studies are very critical and even the use of the intense X-ray flux emitted by synchrotron has not permitted the unambiguous detection of preferential substitution sites by XRD methods [
21] and only an
ad hoc measurements [
24], performed at 170 K, gave a very week evidence that Ti has a preferential tendency to occupy sites T10 and T11. Very recently, a neutron diffraction study [
21] indicates that the most populated sites are T6, T7, T11, with a weaker evidence for T10. Such sites are represented in
Figure 4. In a previous neutron diffraction study [
44] we showed that Si vacancies in defective silicalites do not occur randomly but they are preferentially hosted in the same Si(6), Si(7), Si(11), and Si(10) sites. This striking coincidence allows to speculate that the incorporation mechanism of the Ti atoms in the MFI framework occurs
via the insertion of titanium in defective sites of silicalite. This hypothesis agrees with the well known mineralizing effect that titanium has on the MFI framework and it is supported by several independent spectroscopic data on both TS-1 and defective silicalite [
21,
40].
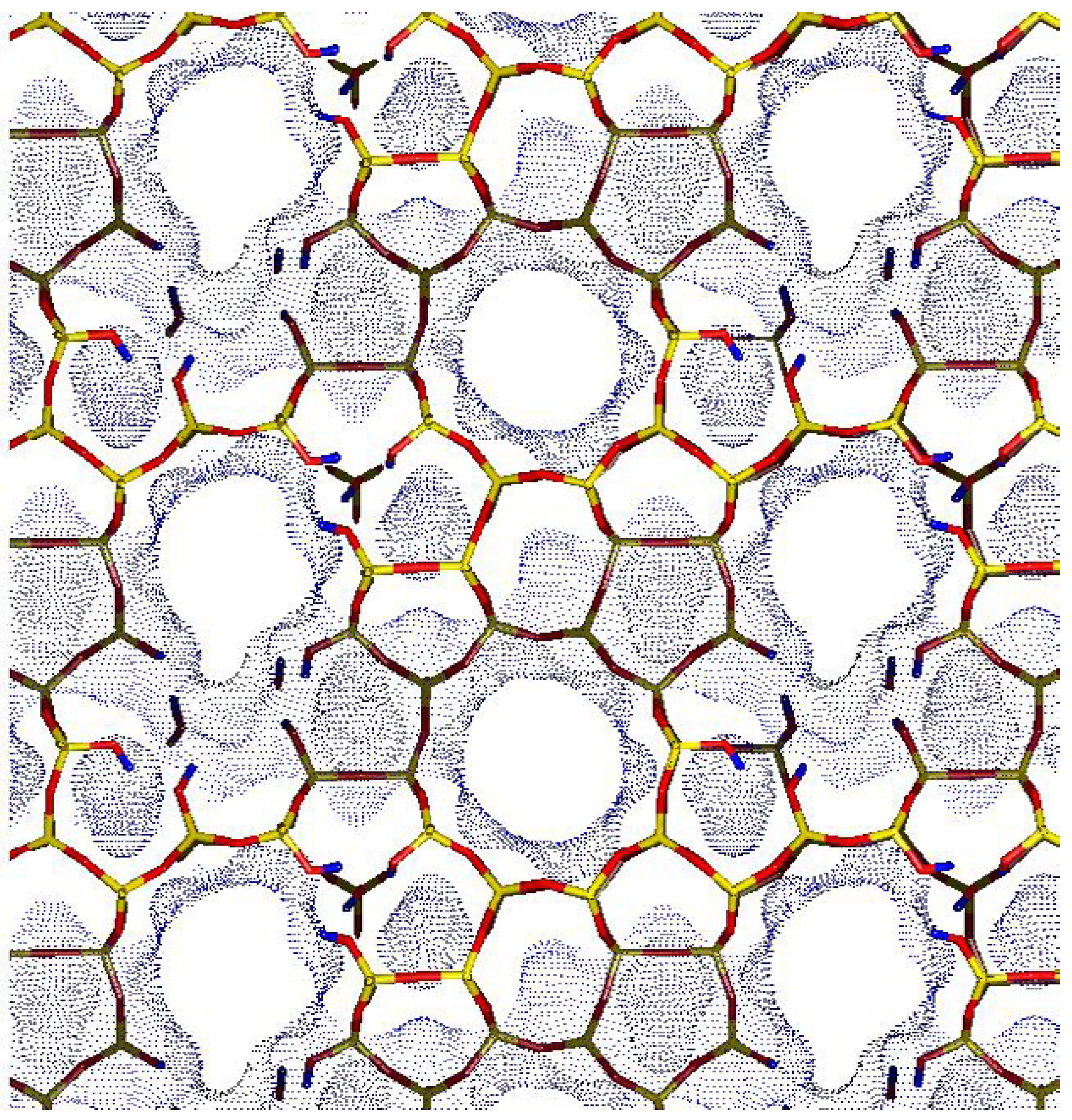
Figure 5 reports a model of a possible hydroxyl nest inside a defective silicalite emerging from our neutron diffraction study [
44]. Since in TS-1, both framework Ti and Si vacancies are present, a reasonable model of TS-1 should be imagined as a merging of the pictures reported in
Figure 4 and
Figure 5. It can so be hypothesized that a Si vacancy could be located in one of the four adjacent T sites of a T site hosting a Ti atom, implying that beside the regular [Ti(OSi)
4] units also defective [HOTi(OSi)
3] units should be considered. This consideration should be considered as a stimulus for future theoretical works aimed to study the geometry and the reactivity of defective [HOTi(OSi)
3] studies.
vii) EXAFS spectroscopy [
22,
34,
35,
36,
37,
40] reports that, within experimental errors, Ti(IV) species are coordinated with 4 oxygen atoms at a Ti-O distance (1.79-1.81 Å) which is much higher than the typical Si-O distance of [SiO
4] units in zeolites (≈1.59-1.60 Å). EXAFS spectroscopy has so explained, on a local ground the origin of the volume unit increase measured with diffraction methods, see point vi). In a couple of EXAFS works, the presence of both regular [Ti(OSi)
4] units also defective [HOTi(OSi)
3] units has been discussed [
39,
40].
It has also been shown that the catalytic activity of Ti site, in several partial oxidation reaction, is related to the fraction of tetrahedral Ti atoms incorporated in TS-1 [
8,
45]. Moreover, theoretical studies have shown that the isomorphous insertion of Ti atoms in the MFI lattice is energetically favored [
46,
47,
48,
49,
50,
51,
52].
Figure 4.
Stick representation of the MFI orthorhombic structure, viewed along the [010] direction: T sites and oxygen atoms have been represented by yellow and red sticks respectively. The preferentially substituted T6, T7, T10 and T11 sites have been evidenced by big blue spheres.
Figure 4.
Stick representation of the MFI orthorhombic structure, viewed along the [010] direction: T sites and oxygen atoms have been represented by yellow and red sticks respectively. The preferentially substituted T6, T7, T10 and T11 sites have been evidenced by big blue spheres.
Figure 5.
Model of a possible hydroxyl nest inside a defective silicalite, where six adjacent T7, T10 and T11 sites have been omitted. This nest is composed by 12 SiOH groups.
Figure 5.
Model of a possible hydroxyl nest inside a defective silicalite, where six adjacent T7, T10 and T11 sites have been omitted. This nest is composed by 12 SiOH groups.
4. The Fe-MFI case study
Insertion of Fe
3+ heteroatoms inside the MFI lattice yields to Fe-silicalite. However, the stability of Fe species in the zeolitic framework is much lower than that of Ti. As a consequence, template burning results in the progressive dislodgment of Fe
3+ species from framework into extraframework positions [
53,
54]. Such migration is far from being a simple and completely understood process. In fact, many factors can influence the final structure and distribution of the potentially active species.
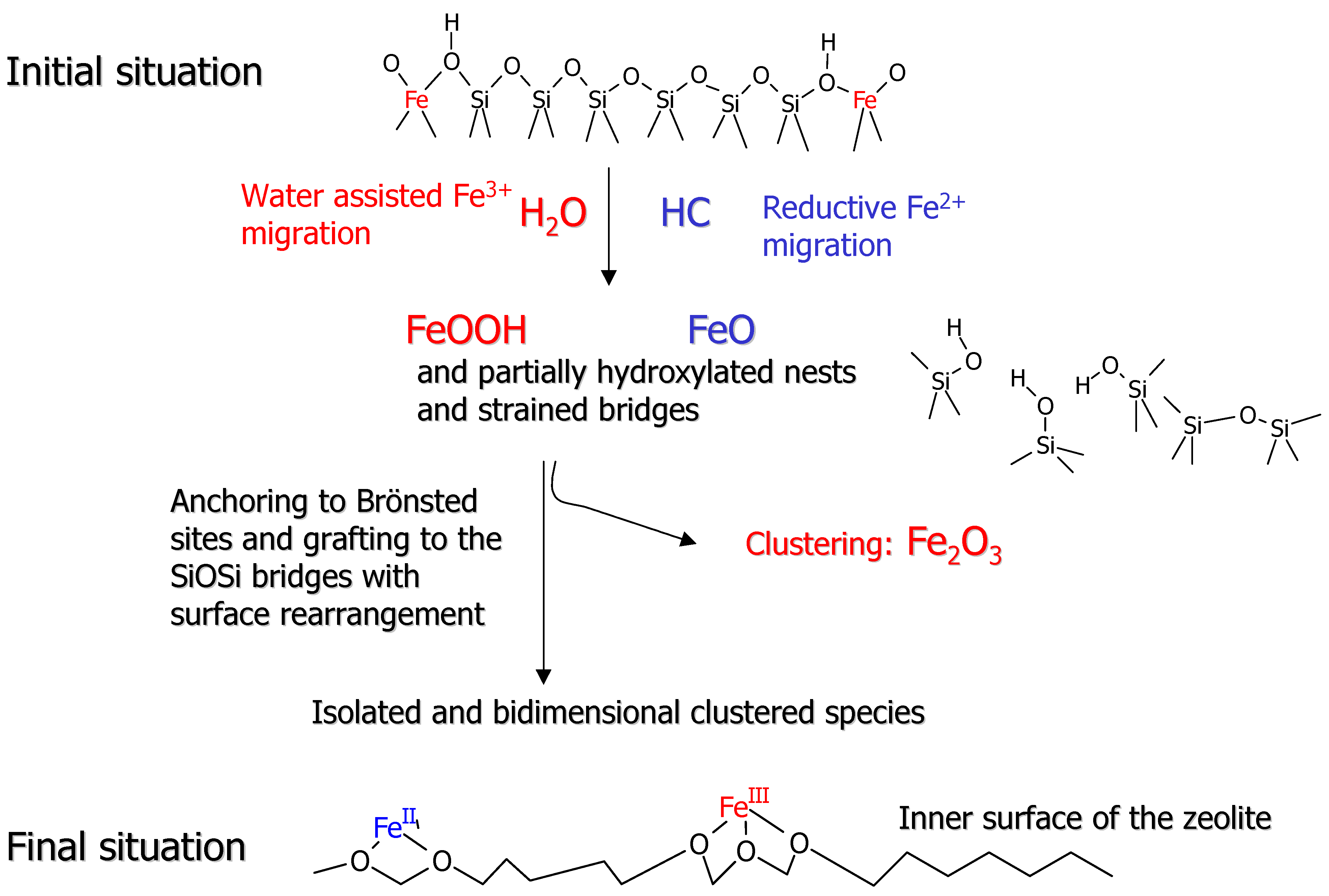
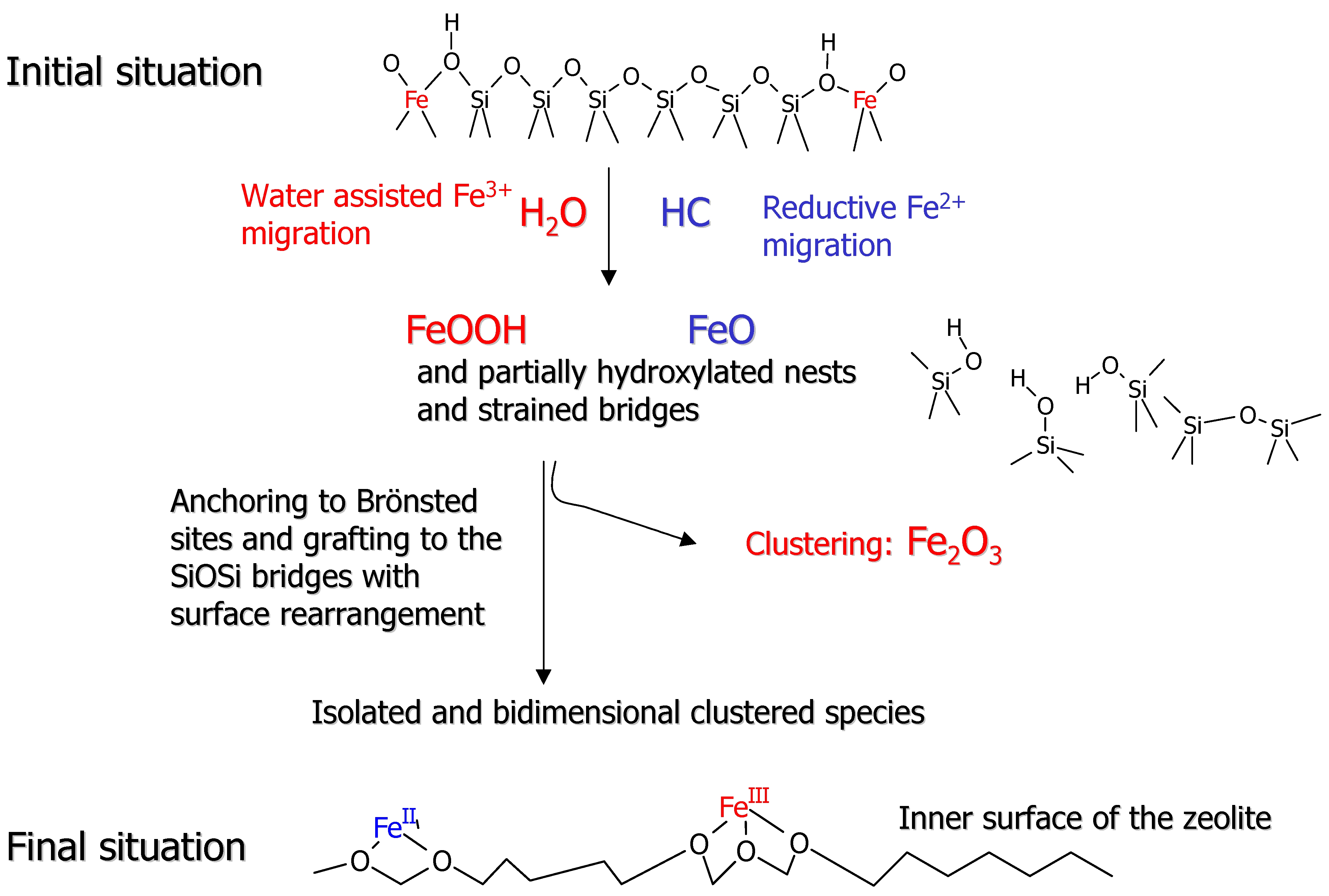
Figure 6 has been conceived to illustrate the remarkable variety of Fe species that can be generated in such a process.
Figure 6 is a pictorial illustration of a pictorial and certainly oversimplified view of the iron dislodgment and migration pathway. The upper part of
Figure 6 reflects the initial situation, where Fe
3+ species are located in the MFI framework forming Fe(OH)Si Brønsted sites. As widely documented for Al [
55,
56,
57] the dislodgment and migration of heteroatoms into extraframework positions in MFI structures is assisted by the presence of traces of water, which favor the breaking of the Si-O and Fe-O bonds and the formation of new SiOSi and SiOFe bridges. We can assume that the dislodgment from the framework position is stepwise, each step being associated with the breaking of one SiOFe bridge and formation of FeOH and SiOH pairs. This process can also be incomplete, giving rise to partially extraframework species. Following this line, it can be reasonably inferred that the total migration of Fe into extraframework position is accompanied by the simultaneous formation of nascent FeOOH species and partially hydroxylated nests or nanocavities which, at high temperature, can eliminate water and form strained SiOSi bridges. It is conceivable that the strained SiOSi bridges formed in the severe temperature treatments can undergo a substantial annealing process [
43]. This migration step is based on the reasonable hypothesis that the dislodgment and migration of Fe
3+ from tetrahedral positions can be favored and brought to completion also by the simultaneous occurrence of red-ox reactions leading to ferrous FeO and/or Fe(OH)
2 species. The redox reactions can be due either to spontaneous loss of oxygen at high temperature or to the presence of hydrocarbons and other carbonaceous impurities [
58,
59] or to both. In samples characterized by a low concentration of iron species, hydrocarbon impurities can play an important role in determining the final oxidation state of the dislodged species. Moreover, the nascent extraframework species can travel along the channels and react with SiOH and strained SiOSi groups or with residual Brønsted Fe(OH)Si sites, resulting in isolated or dimeric grafted species respectively. All species in the presence of traces of water can be mobile so that isolated and dimeric grafted species can be in dynamical equilibrium. It is worth noticing that Fe
2+ and Fe
3+ ions grafted with two and three framework oxygens respectively are very similar to the partially extraframework species obtained in the first stages of the thermal treatments. As a consequence, a dynamical equilibrium between totally and partially extraframework species should be considered. Notice that in all the structures generated following reactions depicted in
Figure 6 the iron centers are chemically linked to 2 or 3 groups: framework SiO
- groups and/or O
2- bridging adjacent iron centers. To prove the heterogeneity of extraframework Fe species we will report EXAFS experiments, while the reductive dislodgment from framework sites will be witnessed by XANES spectroscopy.
Figure 6.
Schematic representation of the different processes involved in the migration from framework into extraframework position.
Figure 6.
Schematic representation of the different processes involved in the migration from framework into extraframework position.
The remarkable success of EXAFS in the characterization of TS-1 catalyst (see refs. [
22,
34,
35,
36,
37,
40] and
vide supra section 3) was mainly related to the fact that Ti active sites in TS-1 are framework species characterized by an homogeneous local environment, which behaves in the same way for all sites upon interactions with reactants. This implies that all Ti sites gives rise to the same contribution which thus adds constructively to the overall EXAFS signal. We will see that Fe-silicalite behaves in a different way.
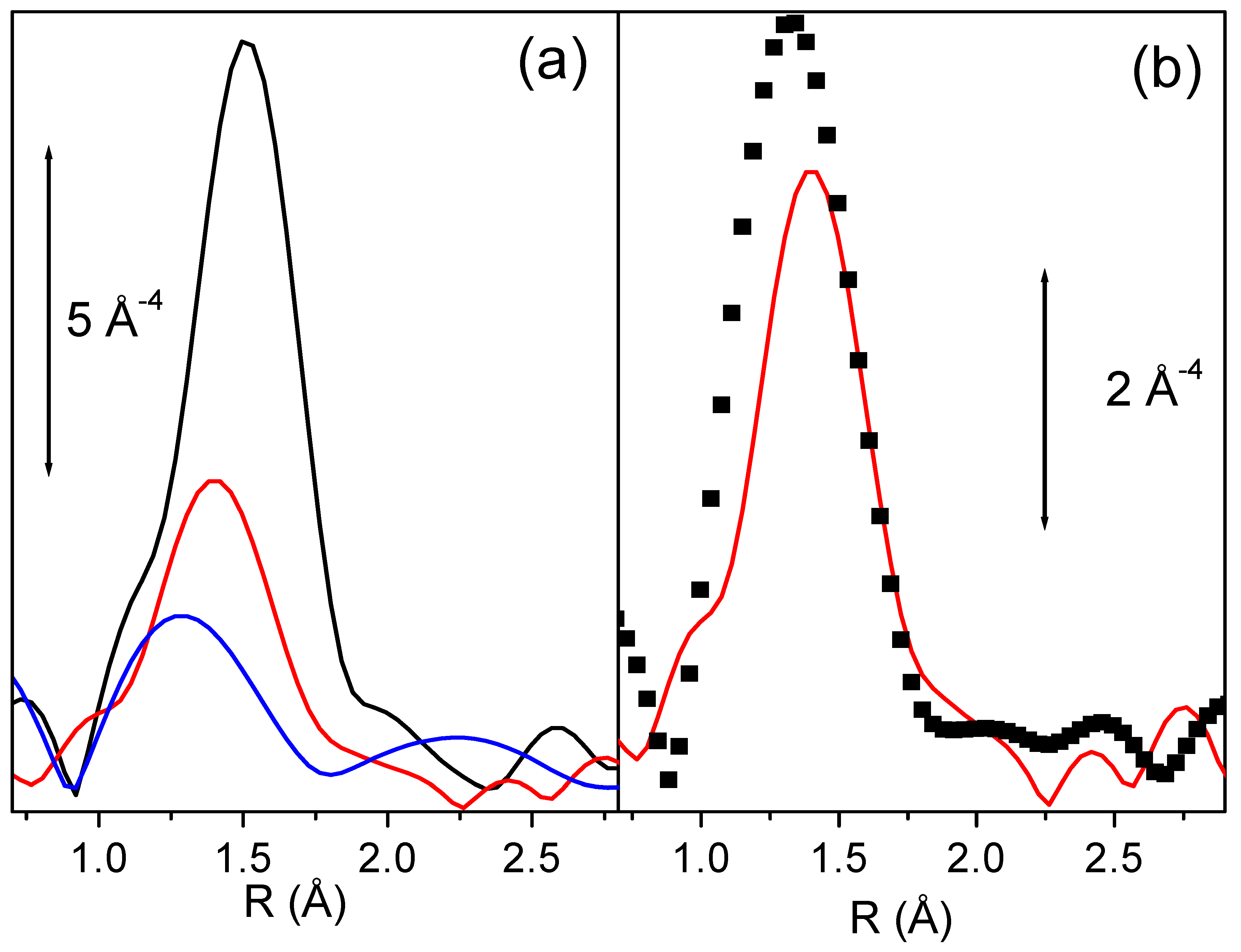
Figure 7a reports the k
3-weighted, phase uncorrected Fourier Transform of the EXAFS functions of Fe-silicalite sample in presence of template and after activation at 773 and 973 K. Even a superficial view on the experimental data allows to remark a dramatic reduction of the EXAFS signal upon activation at increasing temperatures. This impressive phenomenon is related to the high coordinative unsaturation of extraframework species, but has also a second origin. When a fraction of iron atoms migrates into extraframework positions they form a complex variety of isolated and clustered species. Such heterogeneity implies that the local environment of Fe atoms has a consistent spread in both Fe-O bond distances, dynamic Debye-Waller factors and coordination numbers (please note that with O we mean oxygen atoms of both oxidic nanocluster and zeolitic framework at the anchoring site). As a consequence, the EXAFS signal coming from extraframework iron species is affected by a so large Debye-Waller factor (of static origin) to become strongly dumped [
60,
61,
62].
Figure 7.
Part (a): k3-weighted, phase uncorrected Fourier Transform of the EXAFS functions of Fe-silicalite sample in presence of template (black line), and after activation at 773 and 973 K (red and blue lines respectively). Part (b) as part (a) for Fe-silicalite activated 773 K (reported again for sake of comparison, dotted line) and for the same catalyst after subsequent oxidation in N2O at 523 K. Note that the two parts have a different ordinate scale.
Figure 7.
Part (a): k3-weighted, phase uncorrected Fourier Transform of the EXAFS functions of Fe-silicalite sample in presence of template (black line), and after activation at 773 and 973 K (red and blue lines respectively). Part (b) as part (a) for Fe-silicalite activated 773 K (reported again for sake of comparison, dotted line) and for the same catalyst after subsequent oxidation in N2O at 523 K. Note that the two parts have a different ordinate scale.
Figure 7b reports the k
3-weighted, phase uncorrected, FT of the EXAFS spectra collected on Fe-silicalite sample activated at 773 K before and after oxidation with N
2O at 523 K. EXAFS spectroscopy indicates that a considerable fraction of iron ions has undergone an important modification of its coordination sphere, with the insertion of an oxygen ligand at rather short distance [
62]. This ligand should be the adsorbed oxygen atom, produced upon N
2O decomposition, indicated by Panov to be the active site in partial oxidation reactions [
63].
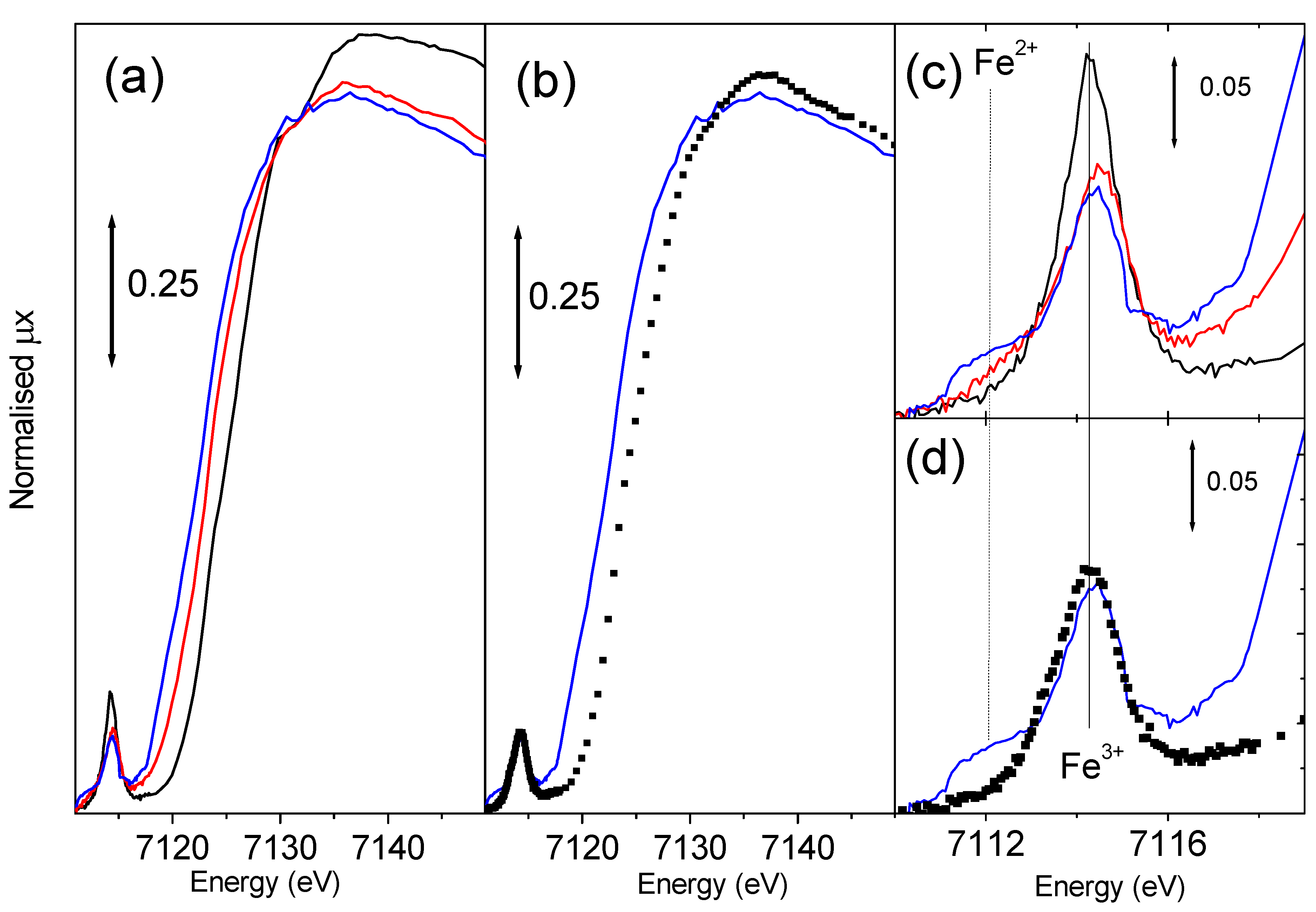
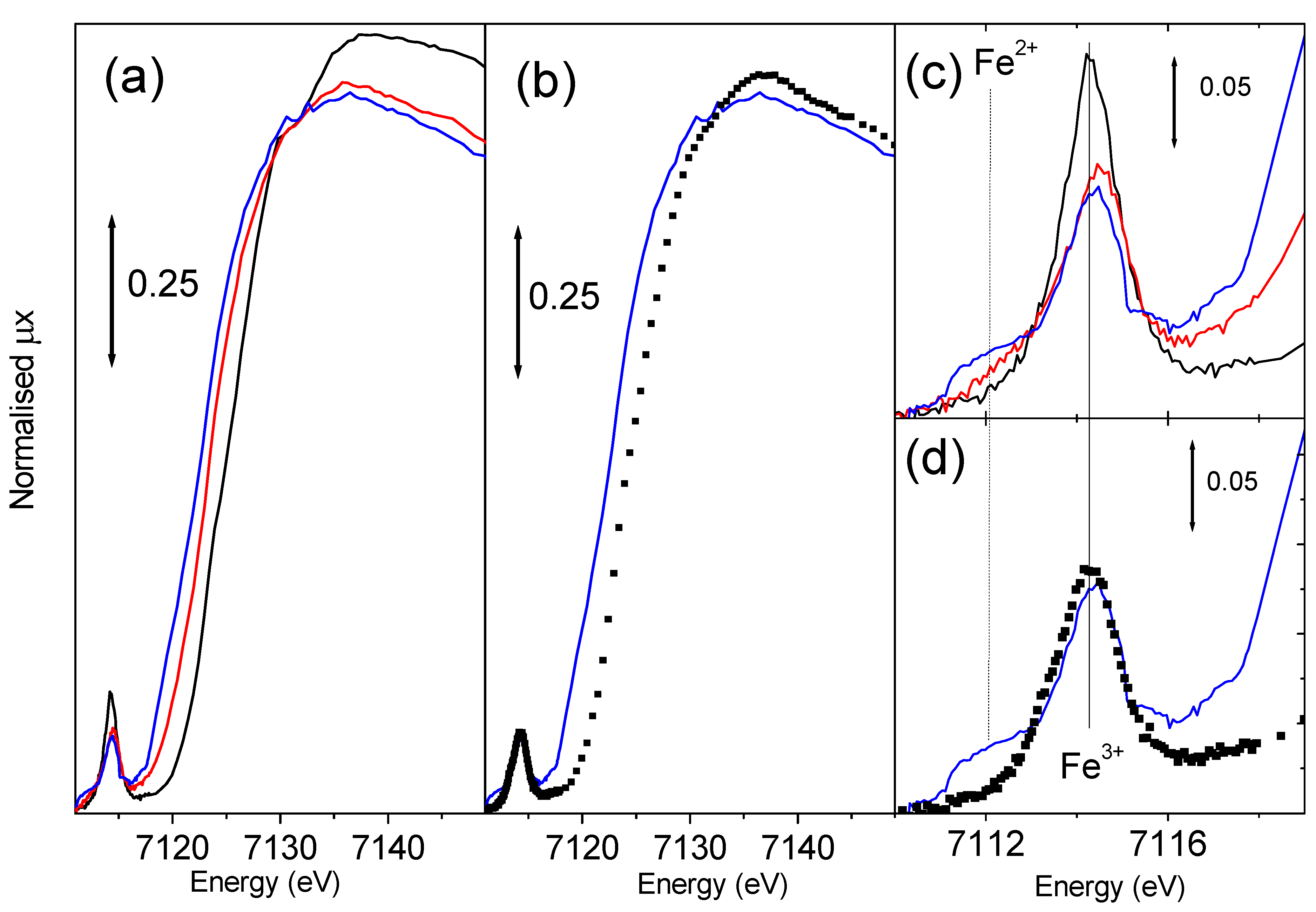
Figure 8.
Parts (a) and (b): as corresponding parts of
Figure 7 for XANES spectroscopy. Parts (c) and (d) reports the magnification of the pre-edge peak of parts (a) and (b) respectively.
Figure 8.
Parts (a) and (b): as corresponding parts of
Figure 7 for XANES spectroscopy. Parts (c) and (d) reports the magnification of the pre-edge peak of parts (a) and (b) respectively.
Being Fe-MFI a red-ox catalyst, the
in situ monitoring of the Fe
3+⇔ Fe
2+ interplay can provide information of fundamental importance. High resolution XANES spectroscopy is one of the most promising techniques to achieve this goal.
Figure 8a reports the effect of template burning at increasing temperature on the Fe-silicalite sample. The shift of the edge position is evident being at 7123.6 eV for the sample measured with template and at 7122.4 and 7120.6 eV for the samples activated at 773 and 973 K respectively. These data confirm that iron in the as prepared sample is present in form of ferric species. After the thermal activation ferric species undergo a progressive reduction to ferrous ones. The intensity of the 1
s → 3
d pre-edge peak (
Figure 8c) is even higher than that of FePO
4 model compound (not reported for brevity) indicating that the local symmetry of Fe
3+ is closer to the ideal tetrahedral than that exhibited in FePO
4. In fact EXAFS told us that in this case we are dealing with 4 equivalent Fe-O bonds at 1.86 Å. The thermal treatment results in a consistent reduction of the 1
s → 3
d peak intensity, demonstrating the evident migration of iron species from the framework tetrahedral positions. In this regard,
Figure 8c shows the appearance of a low energy shoulder in the 1
s → 3
d feature, which intensity progressively increases with increasing activation temperature. The energy position of this new component (around 7111.8 eV) is close to that observed for Fe
2+ compounds [
62]. However, the very large full-width indicates the presence of more than one Fe
2+ species, in agreement with high heterogeneity of extraframework Fe species probed by EXAFS. We notice that the sample with template shows a white line intensity similar to that of FePO
4 (1.31
vs. 1.35 [
62]), much lower than that observed for six-fold coordinated model compounds (which is in the 1.52-1.60 range [
62]), reflecting the four-fold coordination of iron in the as prepared sample. Migration to extraframework positions causes a progressive reduction of the white line intensity: from 1.23 to 1.20 for sample activated at 773 and 973 K respectively. This further reduction of the white line intensity discards the presence of a considerable fraction of iron species in aggregated clusters suggesting the presence of isolated Fe species exhibiting an high coordinative unsaturation. This evidence is confirmed by IR spectroscopy of adsorbed NO, showing that most of the Fe
2+ extraframework species are able to coordinate up to 3 NO molecules [
62,
64].
The effect of a successive oxidation treatment with N
2O at 523 K for 1 h has been investigated by XANES spectroscopy (
Figure 8b and
Figure 8d) for the sample previously activated at 973 K. The edge position shifts back from 7120.6 to 7123.1 eV indicating that an effective oxidation process has been achieved resulting in a nearly complete restoration of ferric species. The same conclusion can be reached by observing the total erosion of the 7111.8 eV component of the 1
s → 3
d pre-edge peak ascribed to Fe
2+ species (
Figure 8d). A similar trend has been observed on the sample activated at 773 K and successively oxidized, in this case the phenomenon is less pronounced because of the low fraction of the Fe
2+ species migrated to extraframework positions. We finally observe that the oxidation process causes the increase of the white line intensity, passing from 1.20 to 1.24. This increase indicates the addiction of a new ligand to the coordination sphere of the extraframework iron species. Also the ability of iron species to coordinated NO molecules has been strongly reduced upon oxidation treatment with N
2O, as monitored by IR spectroscopy [
62].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}