Materials and Methods
and were obtained from Porphyrin Products, Inc. (Logan, UT). Each metalloporphyrin was dissolved in PBS, pH 7.4 and filtered through No.1 filter paper (Whatman, Kent, England) followed by filtration through a 0.2 μm filter (Millipore, Bedford, MA). The Mn concentration for each metalloporphyrin solution was determined by ICP-AAS (Desert Analytics, Inc., Tucson, AZ).
Electronic spectra were recorded on a Shimadzu UV-1601 spectrophotometer. Relaxation measurements were made on a custom-designed variable field T1-T2 analyzer (Southwest Research Institute, San Antonio, TX) at 23 oC. The magnetic field strength was varied from 0.05 to 1.5 T (corresponding to a proton Larmor frequency of 2-61 MHz). T1 was measured by using a saturation recovery pulse sequence with 32 incremental recovery times. T2 was measured by using a Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence of 500 echoes and a time interval of 1 msec between echoes. The relaxivities (relaxation rate per mM concentration) were obtained after subtracting the buffer contribution.
Results and Discussion
A Beer's law plot of the Soret bands indicated no aggregation for either the or the solutions in PBS, pH 7.4. Additionally, no evidence of precipitation was observed for up to at least 6 months.
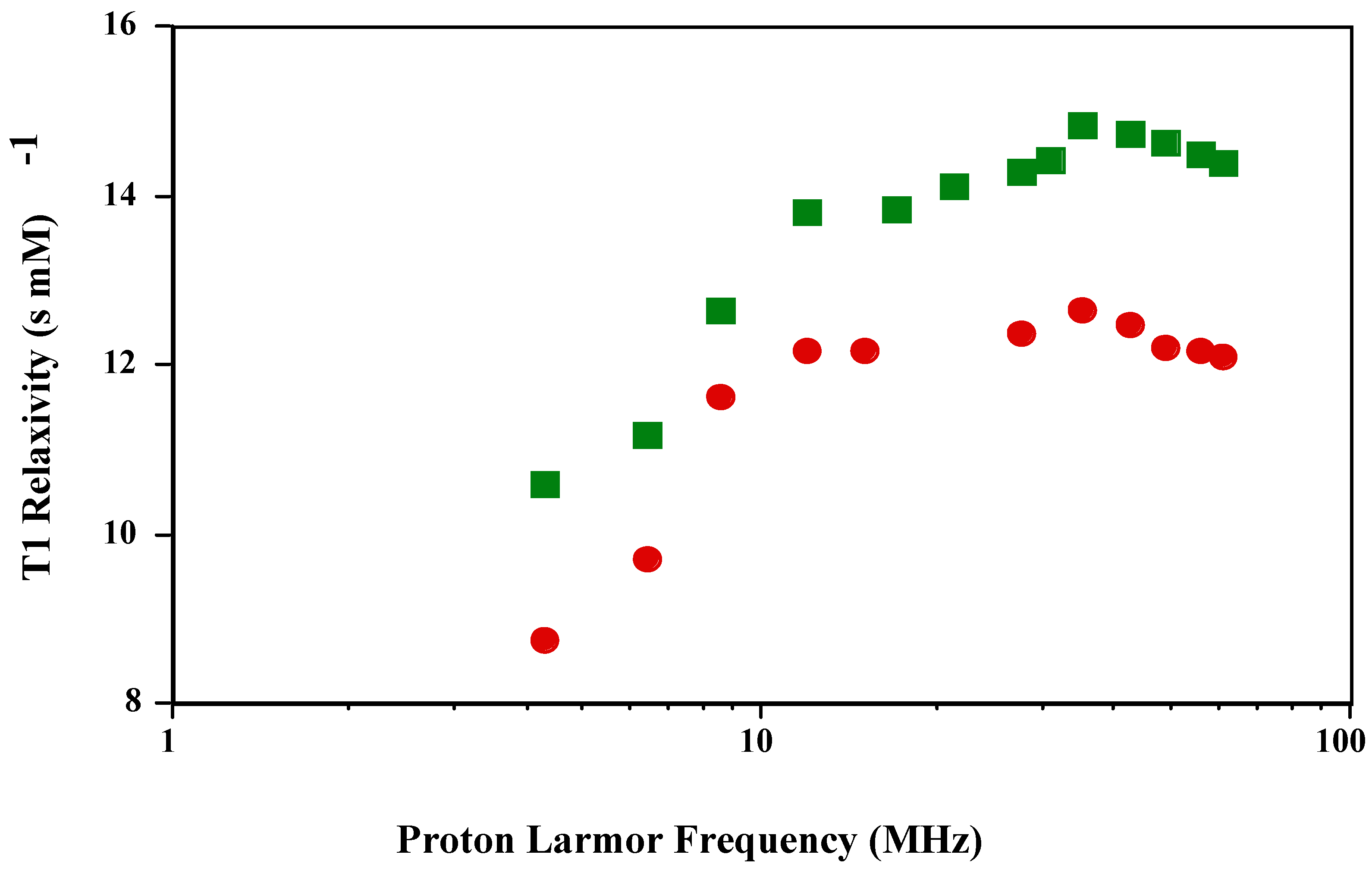
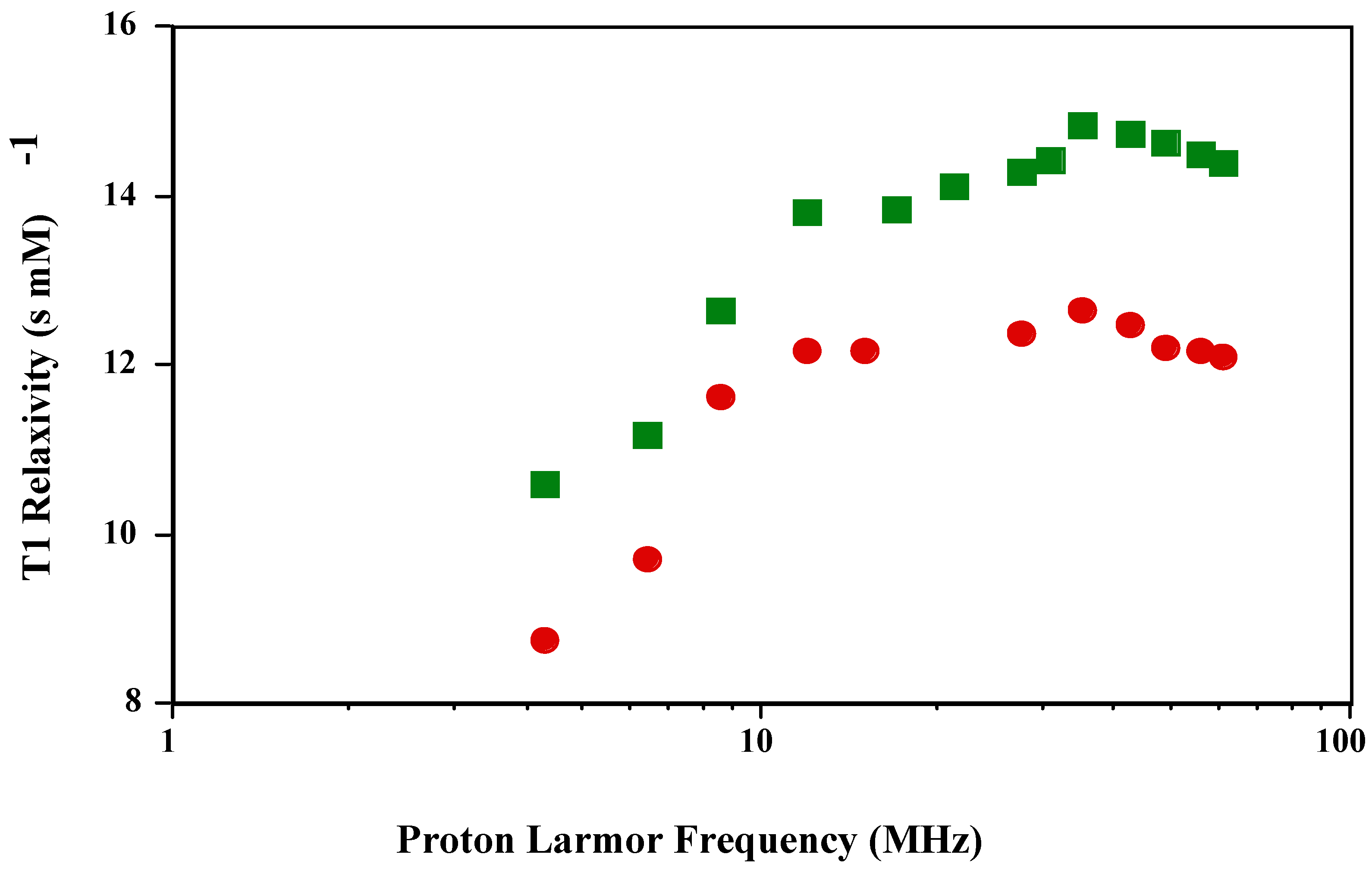
The T1 NMRD profile of
is qualitatively and quantitatively in agreement with previously published reports [
3]. The authors also reported that the T1 NMRD profile of
was the same as MnT(4-C)PP, which has a carboxylate instead of a sulfonato group in the
para position. The T1 and T2 NMRD profile of the new
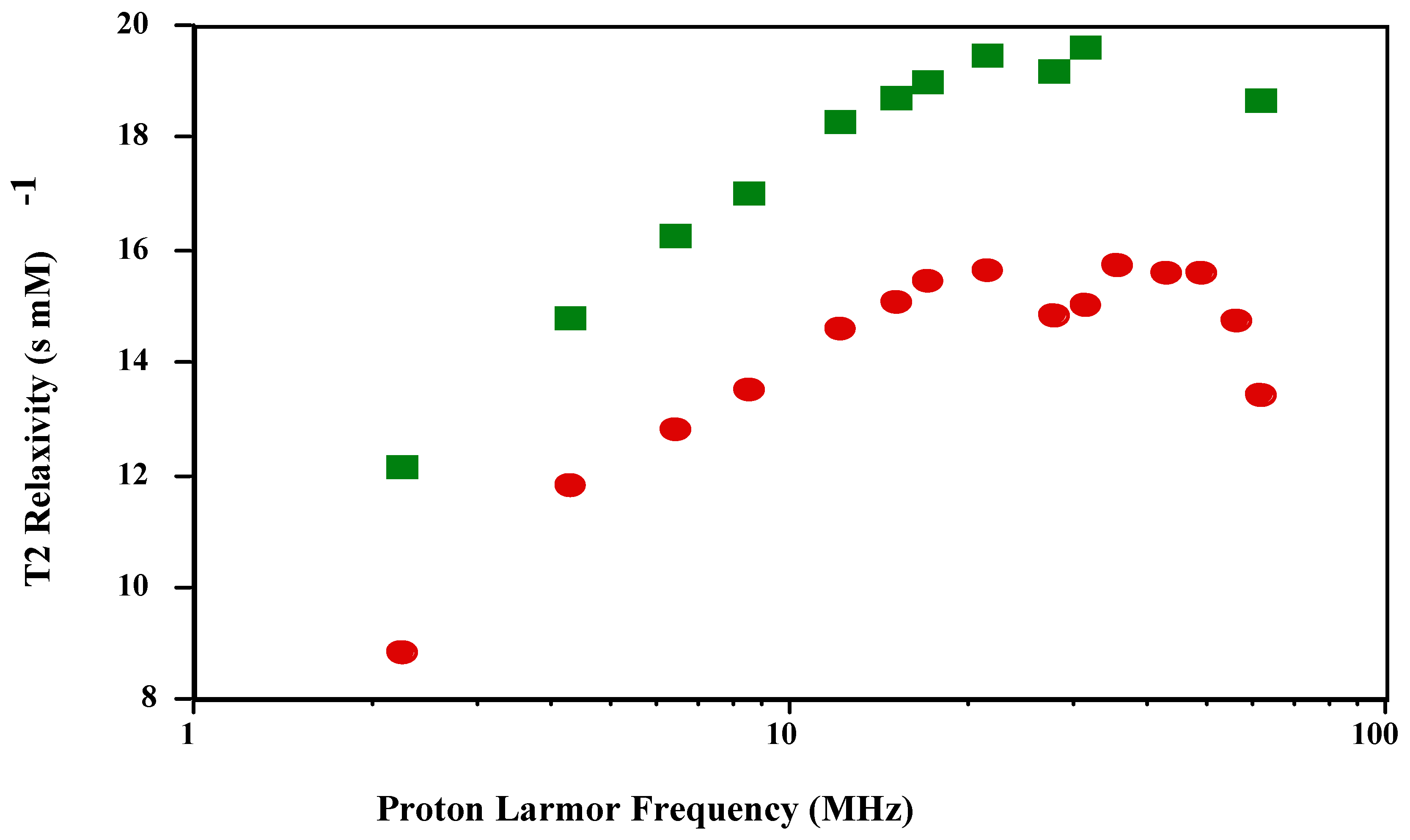
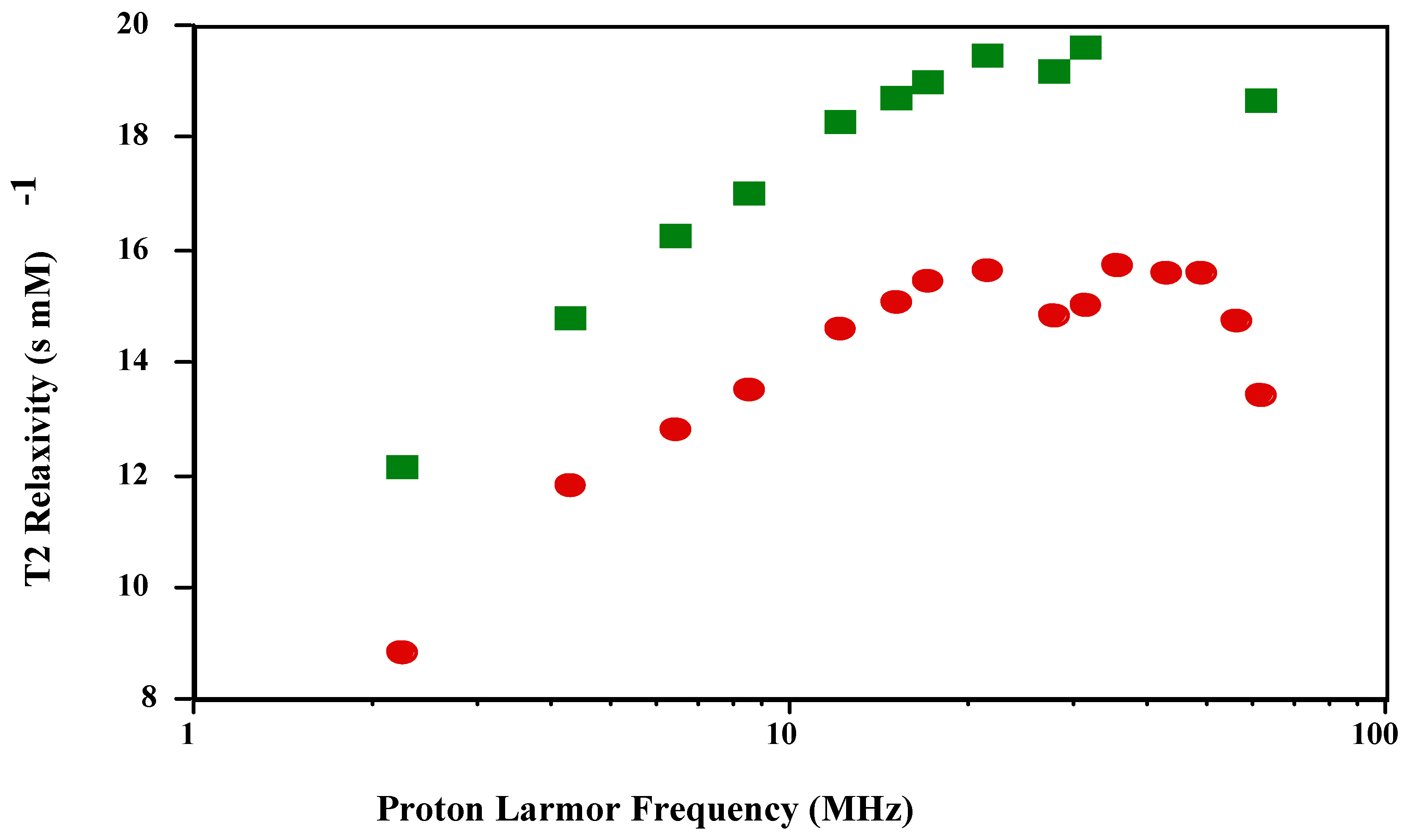
is shown in
Figure 1 and
Figure 2. The shape of the T1 profile for
is similar to that of
and MnT(4-C)PP, but its magnitude is larger by a factor of 1.2. As expected, the T2 NMRD profile parallels the T1 NMRD one.
Figure 1.
T1 NMRD profiles of and in PBS (pH 7.4; 23 oC).
Figure 1.
T1 NMRD profiles of and in PBS (pH 7.4; 23 oC).
Figure 2.
T2 NMRD profiles of and in PBS (pH 7.4; 23 oC).
Figure 2.
T2 NMRD profiles of and in PBS (pH 7.4; 23 oC).
To date,
is the most efficient relaxation enhancement agent of the metalloporphyrins [
2]. Our results herein show that
is yet more efficient. Previous attempts to increase the relaxivities of metalloporphyrins by the judicious placement of various functional groups or atoms have met with limited success [
7] and in at least one report a decrease in the relaxivities was actually observed [
8]. Bryant et al. [
7] have attempted to delocalize the paramagnetic electron spin density in MnTPPS by the covalent attachment of bromines to the ß-pyrrole positions of the porphyrin ring. Although an increase in the relaxivity was reported, the electron-withdrawing bromines decrease the stability of the derivatized MnTPPS with respect to manganese ion release.
The Solomon-Bloembergen-Morgan (SBM) theory and the corresponding mathematical modeling of the NMRD profiles based upon it attempt to quantitate the various parameters contributing to the observed relaxivities. The main contributing parameters come from both inner- and outer-sphere water molecules associated with the paramagnetic metal complex after subtraction of the diamagnetic contribution. The inner-sphere relaxivity results from the number of bound water molecules, the concentration of the metal complex and the distance between the paramagnetic electron spin angular magnetic moment vector and the water proton nuclear angular magnetic moment vector and the correlation time of the metal complex. The correlation time comprises the paramagnetic electron spin relaxation time, the exchange time of the water molecule coordinated to the paramagnetic metal ion with bulk water, and the rotational correlation time of the coupled electron-nuclear angular magnetic moment vector of the paramagnetic electron spin and the proton on the coordinated water molecule, usually taken as the tumbling time of the paramagnetic metal complex. The shortest (fastest) one of these dominates the inner-sphere relaxivity. For the Mn(III) porphyrins, the paramagnetic electron spin relaxation time dominates the correlation time and therefore the relaxivity. The electron relaxation time is frequency dependent and when it disperses there is a characteristic peak in the NMRD profile as shown in
Figure 1. The outer-sphere relaxivity has contributions from the electron spin relaxation time, the distance of closest approach of the water proton nuclear angular magnetic moment vector to the paramagnetic electron spin angular magnetic moment vector taken as the distance of closest approach of the water molecule to the paramagnetic center and the relative translation diffusion coefficient.
Attempts to mathematically model the NMRD profiles using the standard SBM theory were unsuccessful possibly due to zero-field splitting contributions of the manganese coordination environment within the metalloporphyrin [
7,
8,
9]. This type of modeling is a common tool in the effort to develop new compounds with larger relaxivities and that may be applied as contrast media in magnetic resonance imaging[
10]. The inability to do this modeling together with the limited success of empirical methods[
7,
8] makes it necessary to explore new tools that may be used to successfully guide the synthesis of new metalloporphyrins with larger relaxivities. It has been suggested that one such tool is the MEP [
6].
The MEP is an experimentally observed molecular property that is easier to compute with modern computational chemistry methods than to measure [
11,
12,
13]. The MEP is commonly defined by,
where the equation is in atomic units. The first term is the electronic contribution to the electrostatic potential and the second term is the contribution from the atomic nuclei[
11]. Multiple authors have reviewed the role of MEP in molecular reactivity[
11,
12]. Because electrostatic interactions are important in the bonding of water molecules to transition metals and their complexes[
14,
15,
16,
17,
18], Mercier suggested that the MEP may be modified to generate an electrostatic focusing field that would attract water molecules closer to the paramagnet’s spin density[
6]. This closer interaction would also imply a stronger bond. The result would be to increase the relaxivity, not only because the distance between the paramagnetic electron spin angular magnetic moment vector and the water proton nuclear angular magnetic moment vector would be reduced, but also because the number of “bound” water molecules would be increased. These changes would affect both the inner- and outer-sphere mechanism of relaxation. Moreover, an electrostatic focusing field may also increase the residence time of water molecules bound to the inner- and outer-spheres. This effect would increase the outer-sphere contribution to the relaxivity and would have variable effects on the inner-sphere contribution. A more detailed discussion of these issues is found in reference [
6].
It is noteworthy that a simple change from the
para to the
ortho position in the location of the carboxylate group for
increases the relaxivity by 20 % when compared to
. This modest but significant increase is unlikely to result from changes in the electronic configuration of the Mn(III) ion or rotational correlation time. Nonetheless, the change in the location of the anionic functional group has a strong effect on the MEP [
6].
As discussed above and explained by Mercier [
6], the MEP is a marker for the forces responsible for the motion of water molecules around the paramagnet. The fact that a change in relaxivity occurs by a simple pertubation in the molecular geometry that retains the net charge of the complex is significant. This result indicates that the relaxivity in this family of compounds is sensitive to the anisotropy of the electrostatic forces generated by the spatial distribution of the electric charges. Therefore, our results support the suggestion that the MEP can be used as a tool to design compounds with more efficient relaxivities.
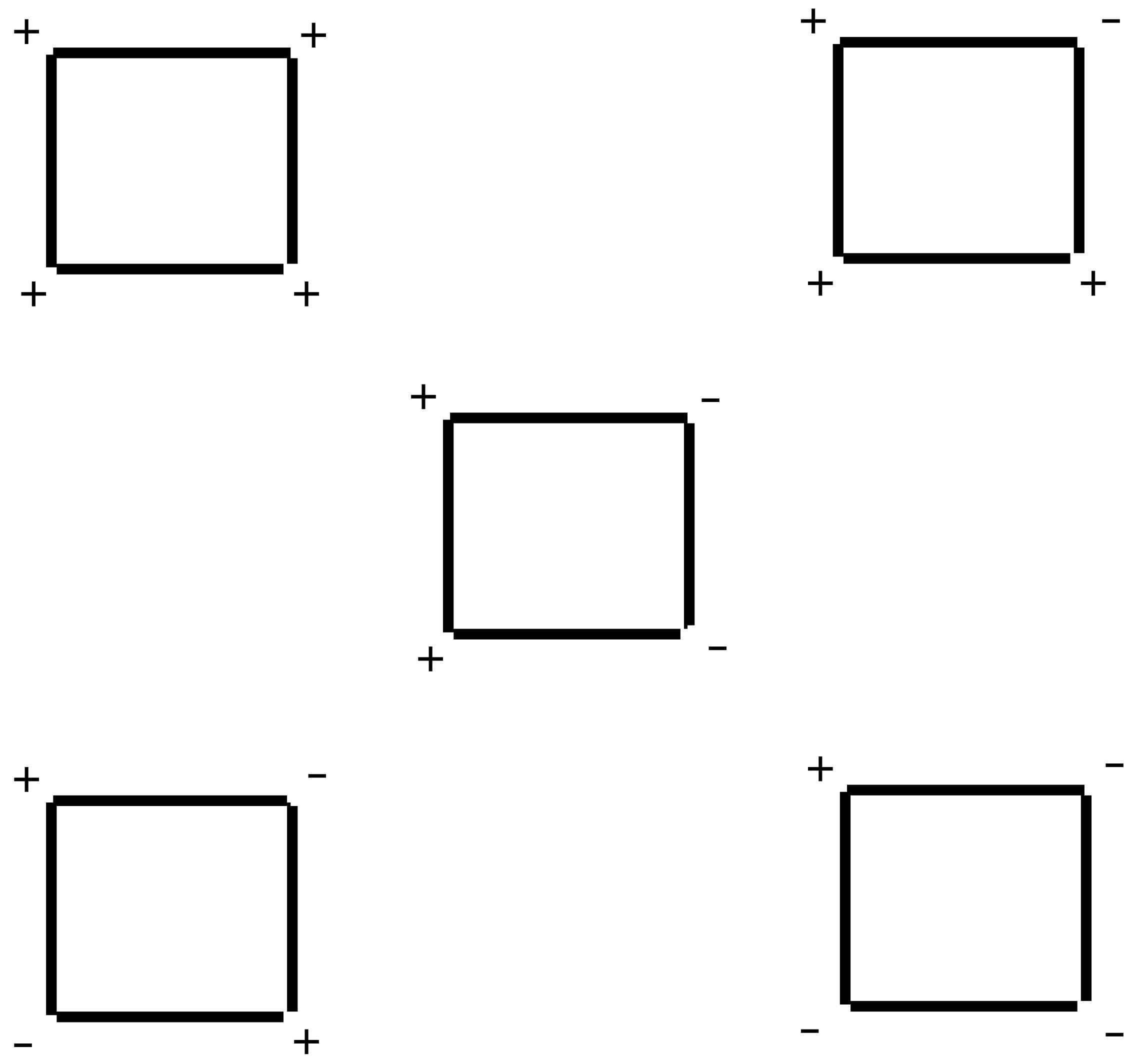
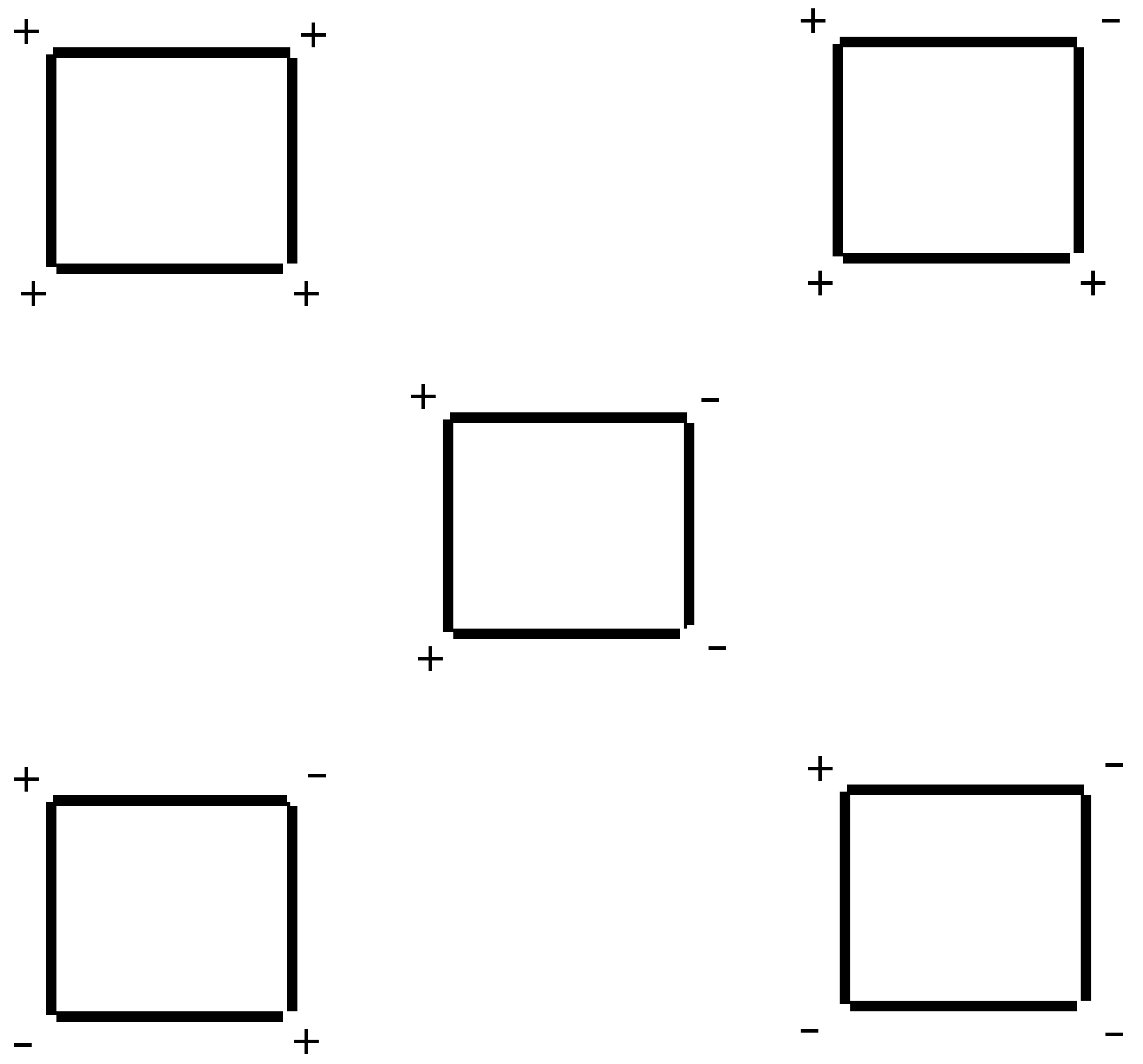
Although the increase in relaxivity is small, our results are encouraging because the formulation of
used in this study constitutes a mixture of conformers. As such, the electrostatic focusing field associated with the MEP is still suboptimal when compared to the one suggested by Mercier [
6]. Because there are hindered rotations around the C
meso-C1 bond, the orientation of the carboxy group in the
ortho position with reference to the plane of the porphyrin ring can vary as shown in
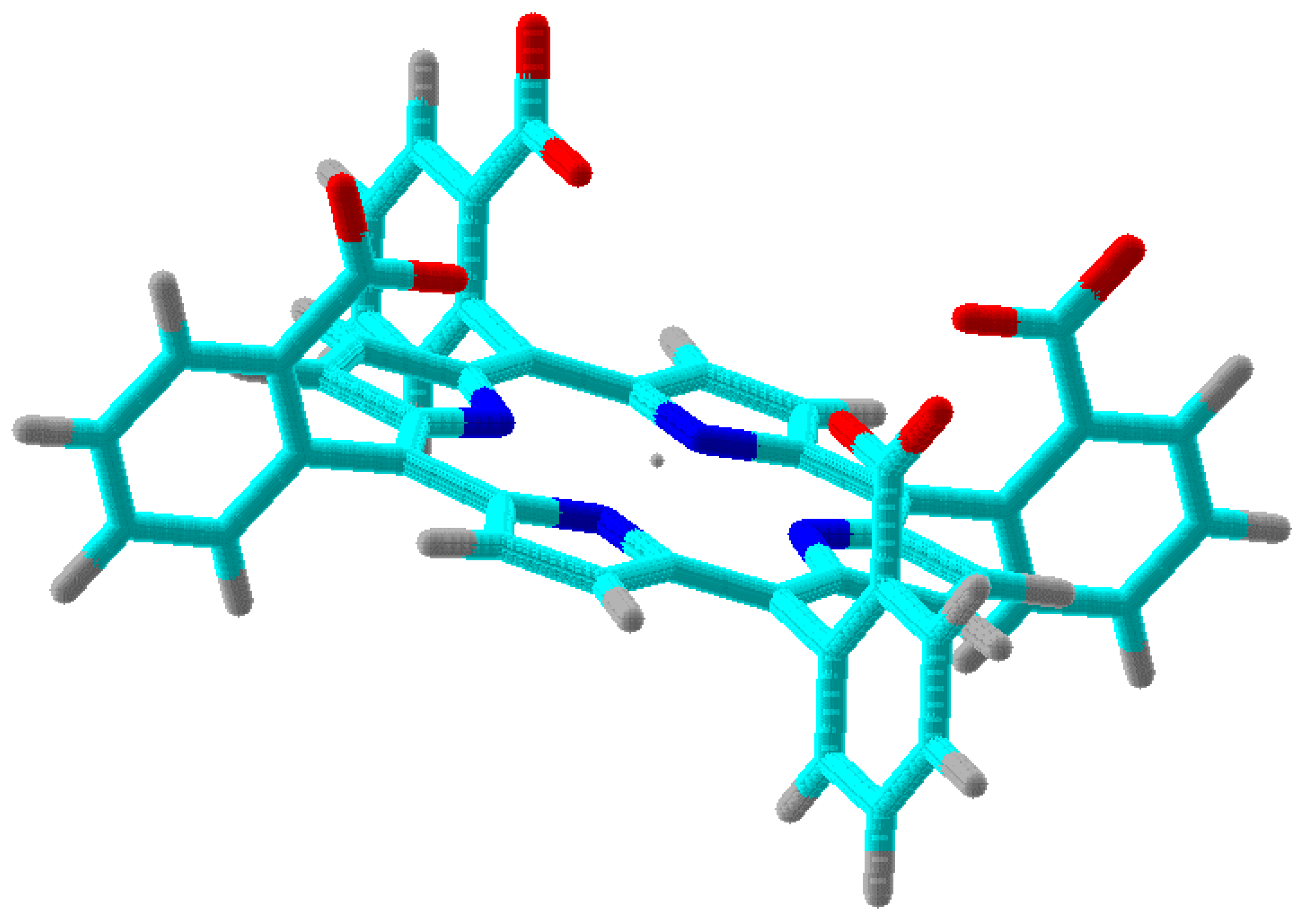
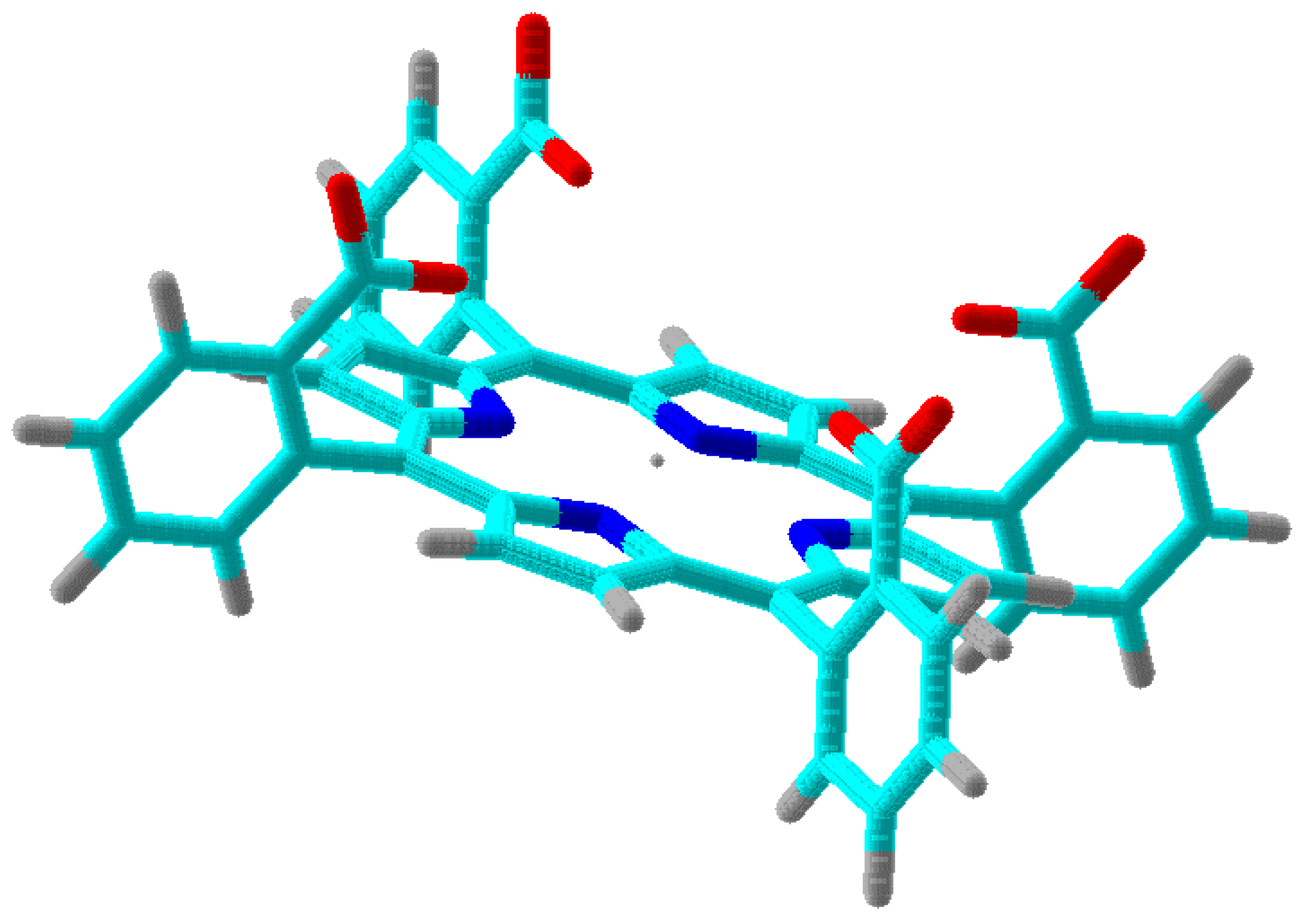
Figure 3. The maximium electrostatic focusing field is expected from conformer (+,+,+,+) which is shown in 3D in
Figure 4. Our results reflect a weighted average of the electrostatic focusing field from all the conformers. Moreover, as discussed previously [
6], the effect of the MEP on the relaxivity should be more dramatic when the rotational correlation time is long. Therefore, there is still room to improve on the modest effect described here.
Figure 3.
Conformers of . "+" reflects a carboxy group above the plane of the porphyrin ring. "-" reflects one below the ring. The Mn(III) ion lies slightly above the plane of the ring.
Figure 3.
Conformers of . "+" reflects a carboxy group above the plane of the porphyrin ring. "-" reflects one below the ring. The Mn(III) ion lies slightly above the plane of the ring.
In summary, we have shown experimentally by NMR dispersion profiles that the manganese porphyrin shows a 20 % increase in relaxation efficiency relative to , supporting the hypothesis that electrostatic forces are relevant to the relaxivity of these types of compounds. The MEP appears to be a useful tool to design new metalloporphyrins with improved relaxivities. We are in the progress of determining the distribution of conformers present in our experiments and isolating the conformers to understand how much we can increase the relaxivity by modifying the MEP. With this work, we hope to further increase the relaxation enhancement of metalloporphyrins by judicious placement of various functional groups on metalloporphyrins. In addition, we intend to explore the in vivo characteristics and toxicity of the new .
Figure 4.
One of the conformers of
shown as a tube structure in 3D. It corresponds to conformer (+,+,+,+) in
Figure 3. Dark blue are nitrogens in the pyrrole rings. Red tubes are oxygens in the carboxy groups. Light blues are carbons and grey tubes are hydrogens. The Mn(III) ion is in the center as an isolated tiny sphere.
Figure 4.
One of the conformers of
shown as a tube structure in 3D. It corresponds to conformer (+,+,+,+) in
Figure 3. Dark blue are nitrogens in the pyrrole rings. Red tubes are oxygens in the carboxy groups. Light blues are carbons and grey tubes are hydrogens. The Mn(III) ion is in the center as an isolated tiny sphere.

{kind=link}
{kind=link}
{kind=link}
{kind=link}