Introduction
In recent years, considerable interest has been focused on the chemotactic peptide formyl-Met-Leu-Phe-OH "fMLPOH" which has been shown to induce lysosomal enzyme release, thereby playing a very important role in the immunological system. Since it was reputed to be a very active agent [
1], various studies have been realized in order to better understand this tripeptide. The influence of terminal groups has been studied, and it has been demonstrated that the esterification of the C-terminal carboxylic acid group does not result in loss of biological activity of molecule [
2]. On the other hand, the replacement of the N-terminal formyl group by tert-butyloxycarbonyl group (Boc) induces a dramatic loss of activity [
3].
In an attempt to produce synthetic agents that are more active and more resistant to enzymatic hydrolysis, several modifications have been undertaken [
4,
5,
6,
7,
8]. The replacement of the Met by the thiomethionine residue (Met
S) induces a dramatic loss of activity [
4]. The comparative study of the formyl-Met-Leu-Phe-OMe and formyl-Met
S-Leu-Phe-OMe has shown that an active chemotactic peptide must have the formyl group free of any intramolecular interaction in order to be available for the formation of the complex with the receptor [
5].
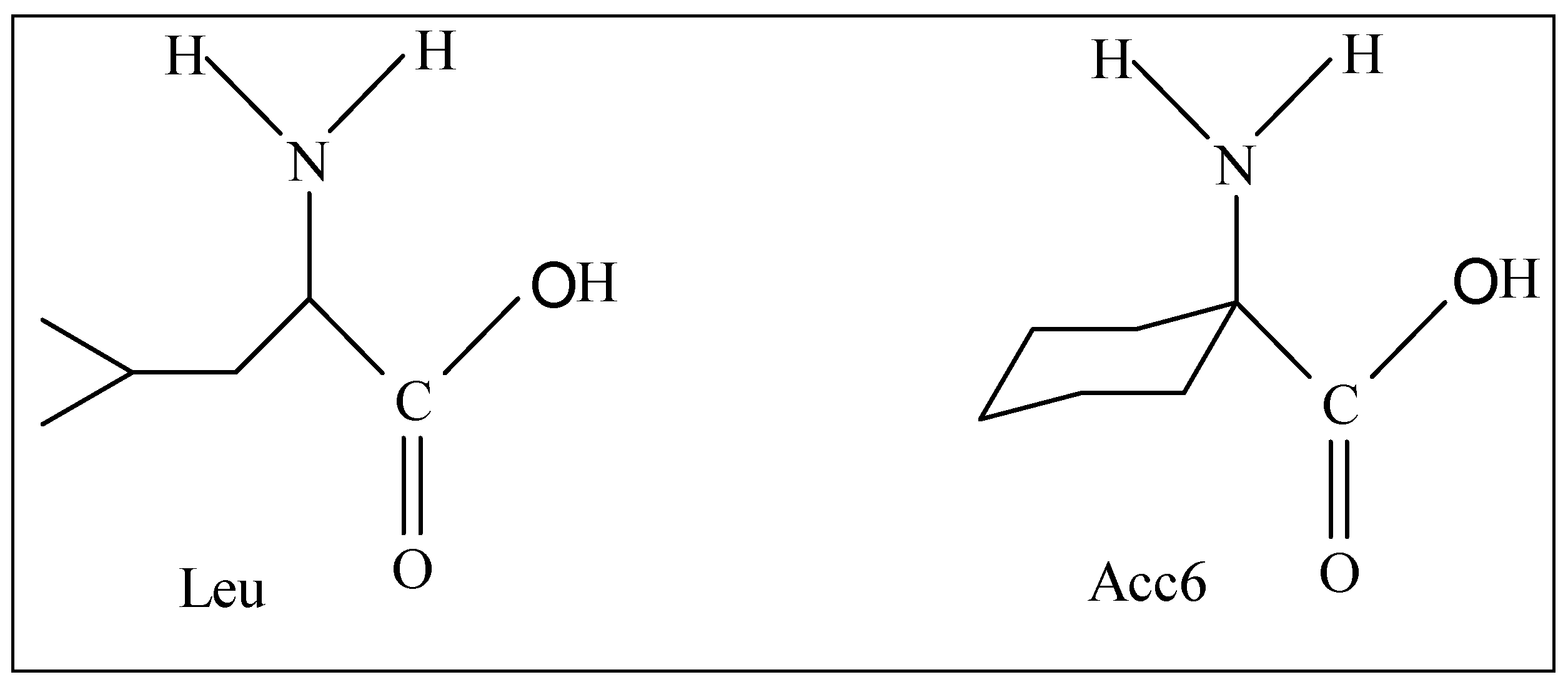
Among the modifications made on the tripeptide formyl-Met-Leu-Phe-OMe, we can quote the substitution of the Leu residue by the α,α-disubstituted amino acids such as Acc6 (l- aminocyclohexane-1-carboxylic acid).
Figure 1.
Structures of amino acids Leu and Acc6.
Figure 1.
Structures of amino acids Leu and Acc6.
This residue was the subject of a considerable number of conformational studies [
9,
10]. The presence of a α,α-disubstituted carbon atom in this amino acid confers upon it the property to have a considerable steric effect, which can then impose significant constraints on the orientation of the peptide backbone. The use of this residue in a given peptide will facilitate the determination of the conformation adopted during the interaction with the receptor, as well as the search of pharmacophore groups.
To measure the activity of formyl-Met-Leu-Phe-OMe and formyl-Met-Acc6-Phe-OMe, their ability to induce the release of lysosomal enzymes in rabbit neutrophils was used. These measurements, which were undertaken by Sukumar's group, showed that the constrained peptide formyl-Met-Acc6-Phe-OMe is approximately six times more active than the parent peptide formyl-Met-Leu-Phe-OMe [
1].
The conformational study, that was carried out by
1H-NMR, IR, and X-rays on these two peptides and other formyl-Met-Leu-Phe-OMe analogs containing other α,α-disubstituted amino acids, has not been able to give a common structure that can explain the biological activity of the chemotactic peptides [
11,
12,
13]. This can be explained by the fact that the analyses by the various experimental methods are carried out in media and environments that are different to those in which these peptides exert their biological function. Consequently, the resulting structures are not necessarily the active structures.
Using a theoretical method, the present article is interested in the comparative conformational analysis of formyl-Met-Leu-Phe-OMe and formyl-Met-Acc6-Phe-OMe. The main objective of this study is to find the active conformation of chemotactic peptides.
Method
The method used in this study is called PEPSEA (PEPtidic SEArch). It was developed in the structural chemistry laboratory of the Sherbrooke University [
14]. This approach is based on the fact that the structural, thermodynamic and statistical properties of a molecular system can be deduced only from a population presenting its conformational space. The principle of PEPSEA consists of generating a population of conformations which characterize a particular peptidic sequence. Rather than striving for global minima, populations of conformers are randomly generated, and their energy is minimized. A statistical analysis can be applied upon these populations to deduce the thermodynamic and structural properties of the peptide under investigation. This new approach is applied with the PEPSEA program.
The force field used by the PEPSEA program to compute the conformational energy is ECEPP/2 "Empirical Calculation Energy Program for Peptide" [
15]. This force field uses rigid geometry to represent the amino acid residues of a polypeptidic chain. The conformational energy function is given by the sum of the electrostatic term
Eele, 12-6 Lennard-Jones term
ELJ, and hydrogen-bond term
Ehb for all pairs of atoms in the molecule together with the torsion term
Etor for all torsion angles.
The PEPSEA program uses the specific parameters of each residue (atomic coordinates, geometrical and energy parameters...) to describe the geometry of a peptidic molecule. The force field ECEPP/2 possesses the parameters of 26 amino acid residues and of terminal protecting groups commonly found in proteins. However, the Acc6 residue is not included in the database, so it is necessary to calculate its parameters and integrate them in the force field ECEPP/2. The atomic partial charges for this particular residue are computed by CNDO calculation [
15].
As all endogenous peptides, the tripeptides under investigation in this study are constituted by the sequence of amino acids, all in L configuration.
Results and Discussion
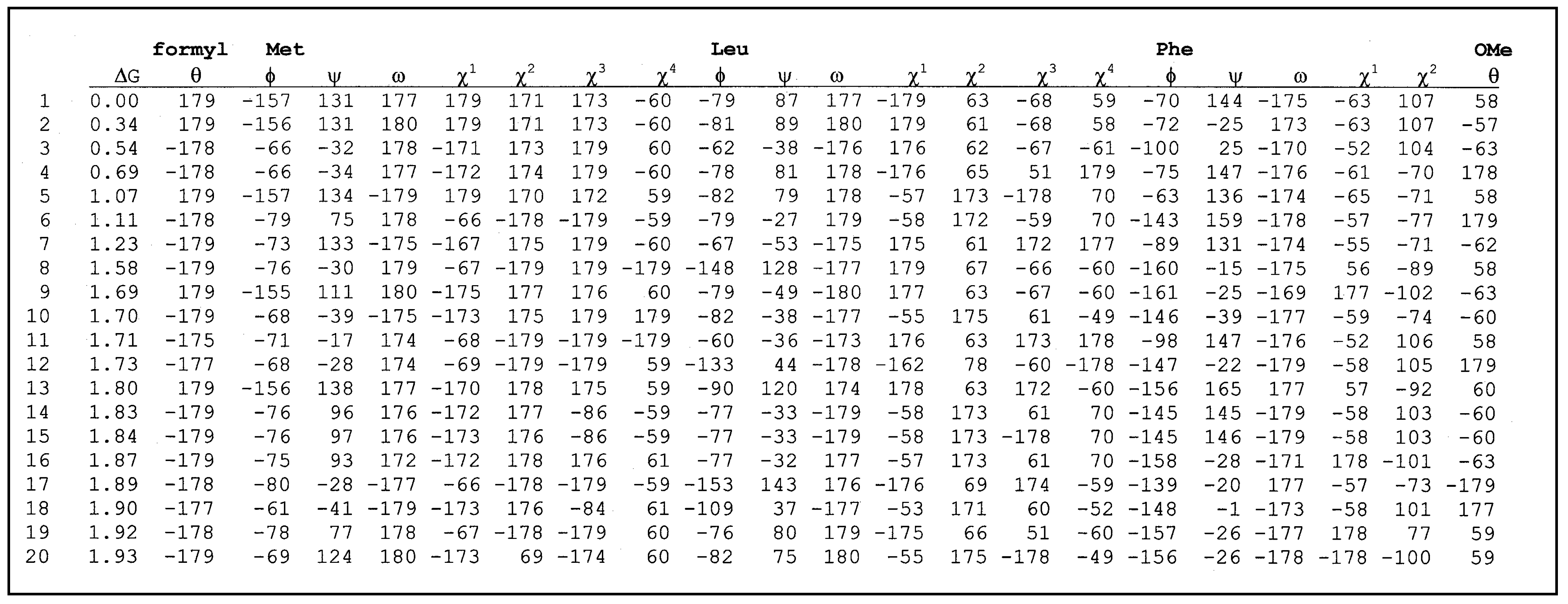
Table 1 and
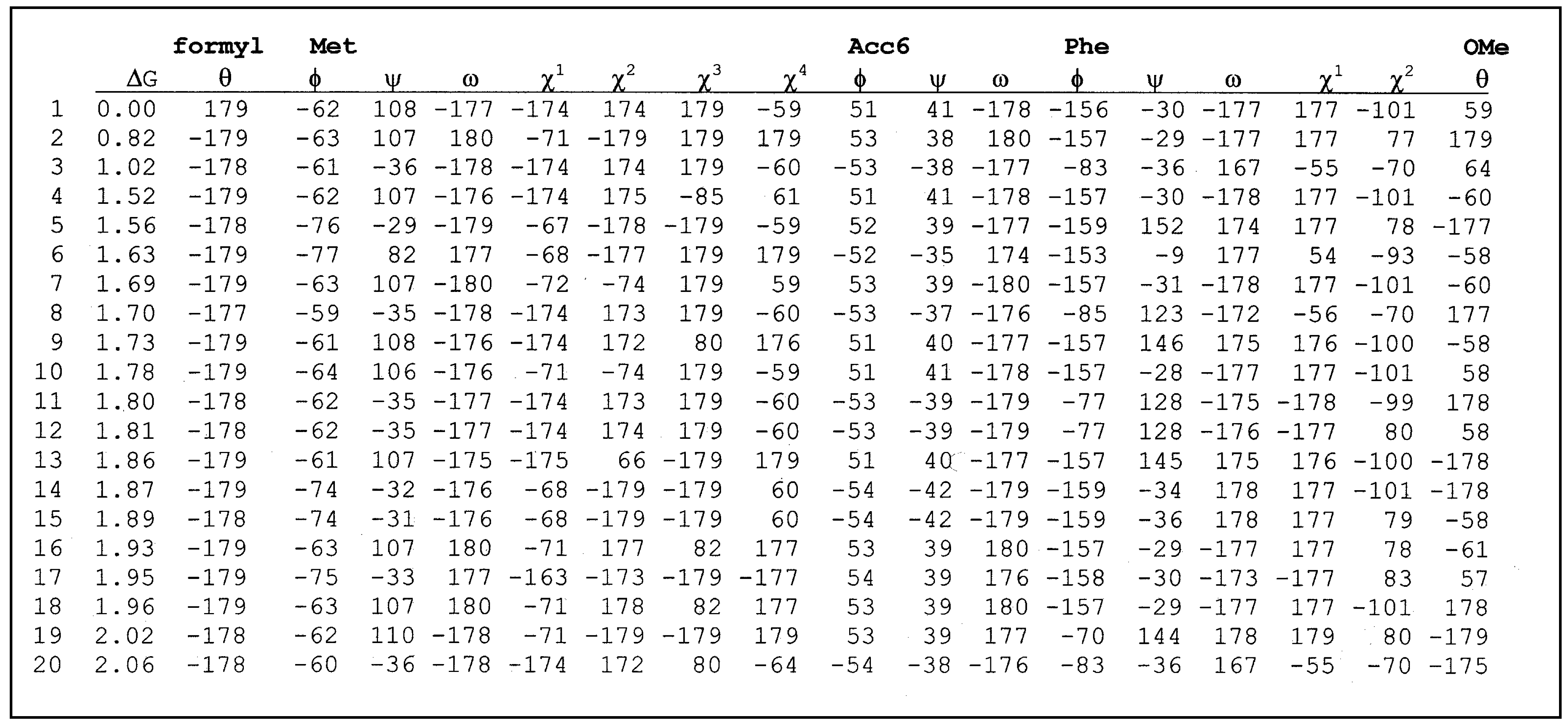
Table 2 give the conformational characteristics of the twenty most stable conformers obtained after the second minimization for each peptide. These conformers are classified by order of increasing relative free energy. For each conformation, we find the relative free energy Δ
G calculated for
T = 300 K, and the relative conformational energy Δ
E. The structural characteristics of each conformer are given by indicating the presence or not of intramolecular hydrogen bonds between the different donors and acceptors. The torsional angles for the parent peptide as well as the constrained one are listed in
table 3 and
table 4 respectively.
Table 1.
Conformational characteristics of formyl-Met-Leu-Phe-OMe a.
Table 1.
Conformational characteristics of formyl-Met-Leu-Phe-OMe a.
| Conf. | Relative free energy ΔG b (kcal/mol) | Relative conformational energy ΔE c (kcal/mol) | Formyl | Met | Leu | Phe |
|---|
| 1 | 0.00 | 0.00 | | CO | | NH |
| 2 | 0.34 | 0.07 | | CO | | NH |
| 3 | 0.54 | 0.38 | CO | | | NH |
| 4 | 0.69 | 0.60 | | CO | | NH |
| 5 | 1.07 | 0.39 | | CO | | NH |
| 6 | 1.11 | 1.41 | CO | | NH | |
| 7 | 1.23 | 1.00 | CO | | NH | |
| 8 | 1.58 | 1.37 | | | | |
| 9 | 1.69 | 2.37 | | | | |
| 10 | 1.70 | 0.92 | CO | | | NH |
| 11 | 1.71 | 1.40 | CO | | | NH |
| 12 | 1.73 | 1.73 | CO | | | NH |
| 13 | 1.80 | 0.99 | | CO | | NH |
| 14 | 1.83 | 1.53 | CO | | NH | |
| 15 | 1.84 | 1.53 | CO | | NH | |
| 16 | 1.87 | 1.43 | CO | | NH | |
| 17 | 1.89 | 1.83 | | | | |
| 18 | 1.90 | 0.66 | | | | |
| 19 | 1.92 | 2.52 | CO | CO | NH | MH |
| 20 | 1.93 | 2.11 | CO | CO | NH | NH |
The conformational analysis of the twenty most stable conformers of the parent peptide formyl-Met-Leu-Phe-OMe (
Table 1) shows that it can adopt varied conformational structures, which can be distributed into four classes:
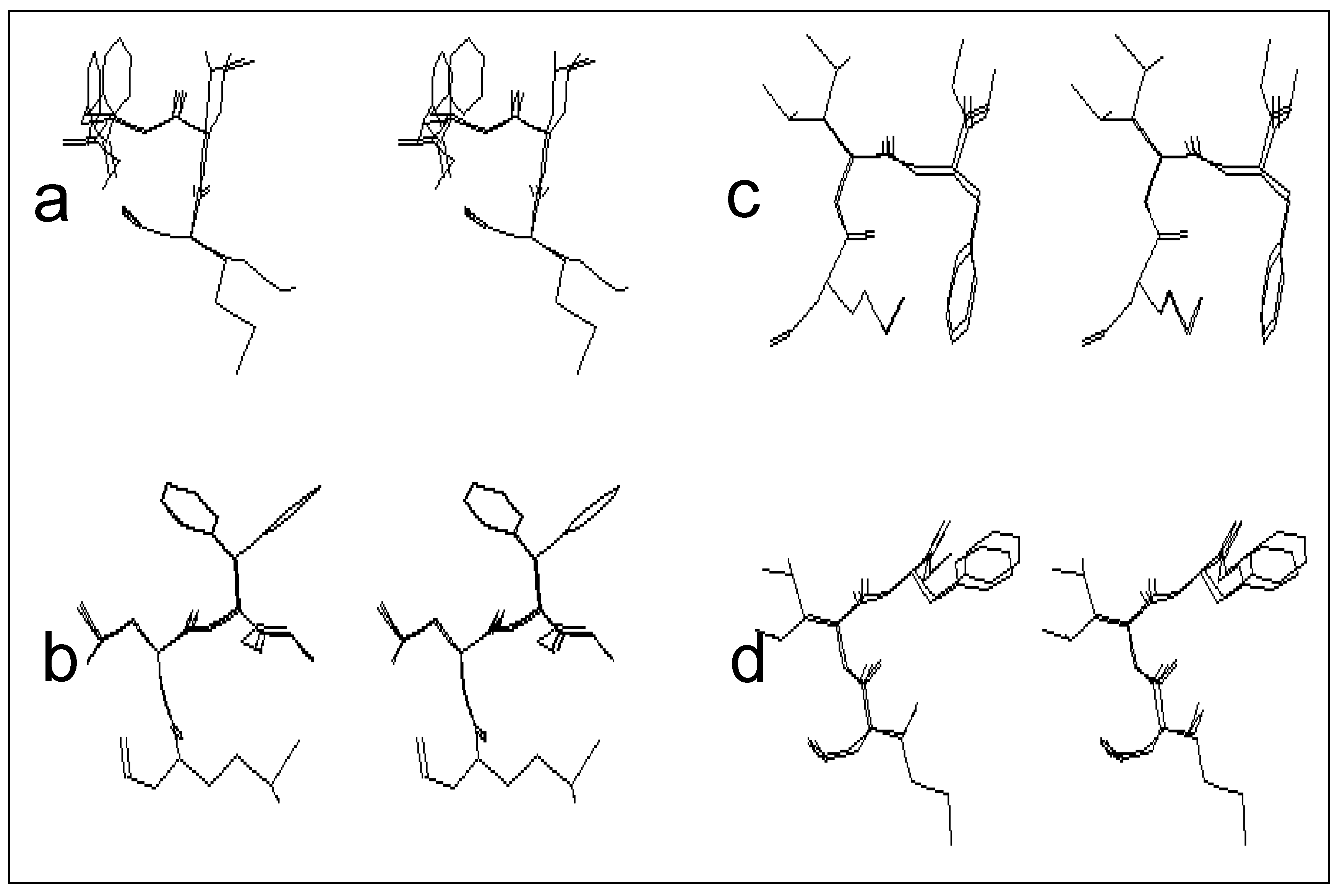
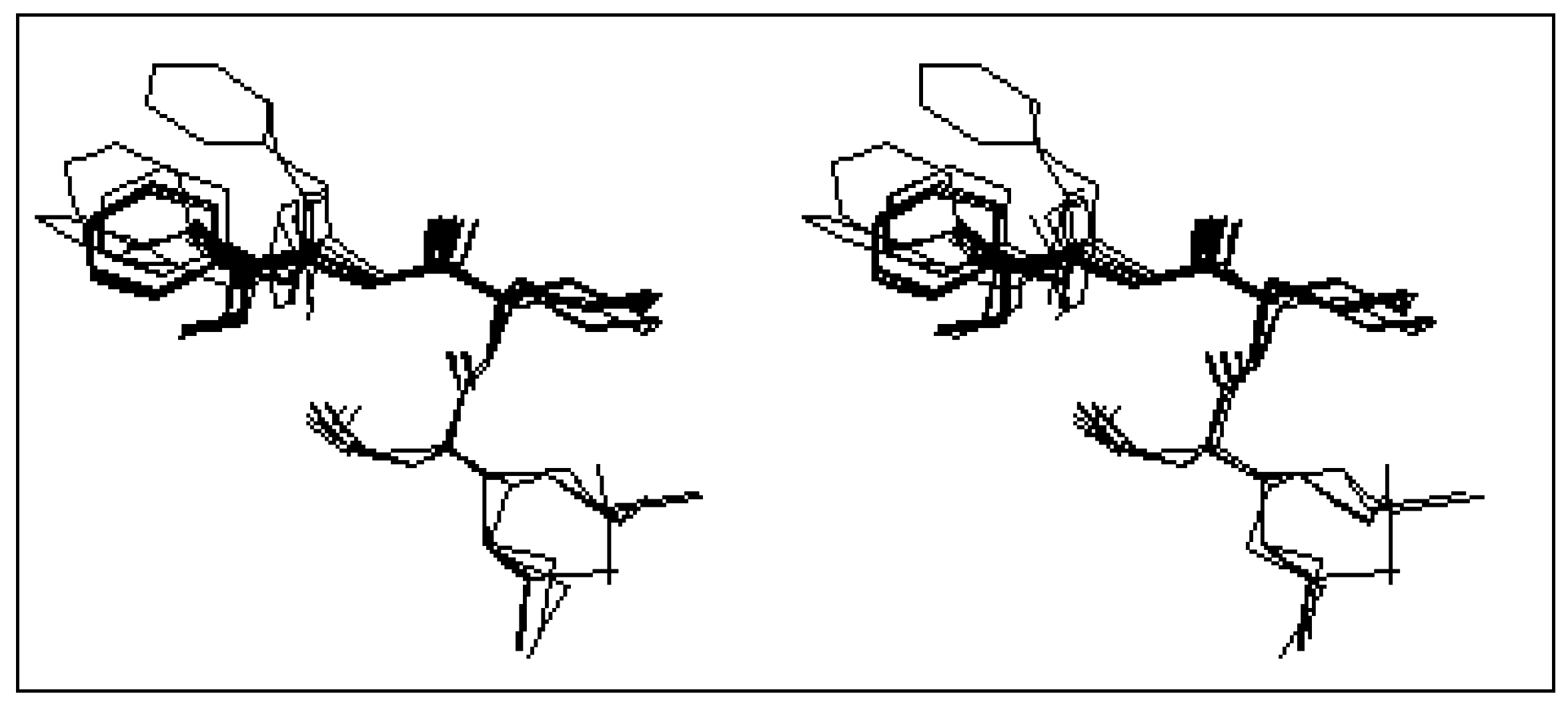
The first class is that of the conformers characterized by the presence of the β turn structure centered on Met and Leu, and it can be represented by four conformers (conformers 3, 10, 11 and 12). Such a structure is stabilized by an intramolecular hydrogen bond including the CO group of the formyl and NH group of Phe.
Figure 2-a gives a stereoscopic superposition view of the four conformers belonging to this group.
The second class includes five conformers characterized by the presence of a γ turn centered on Met, and stabilized by an intramolecular hydrogen bond involving the CO group of the formyl and NH group of Leu. The stereoscopic superposition view of these five conformers is given in
figure 2-b.
The third class, which includes five conformers, is characterized by conformations adopting a γ turn centered on Leu, and stabilized by an intramolecular hydrogen bond, implying the CO group of Met and NH group of Phe.
Figure 2-c gives the stereoscopic superposition of these five conformers.
The fourth class gathers structures in a double γ turn (a γ turn centered on Met and a γ turn centered on Leu at the same time), and includes two conformers of formyl-Met-Leu-Phe-OMe. The stereoscopic superposition view of both conformers of this class is presented on the
figure 2-d.
Figure 2.
Stereoscopic superposition view of conformers of different classes obtained in the case of formyl-Met-Leu-Phe-OMe.
Figure 2.
Stereoscopic superposition view of conformers of different classes obtained in the case of formyl-Met-Leu-Phe-OMe.
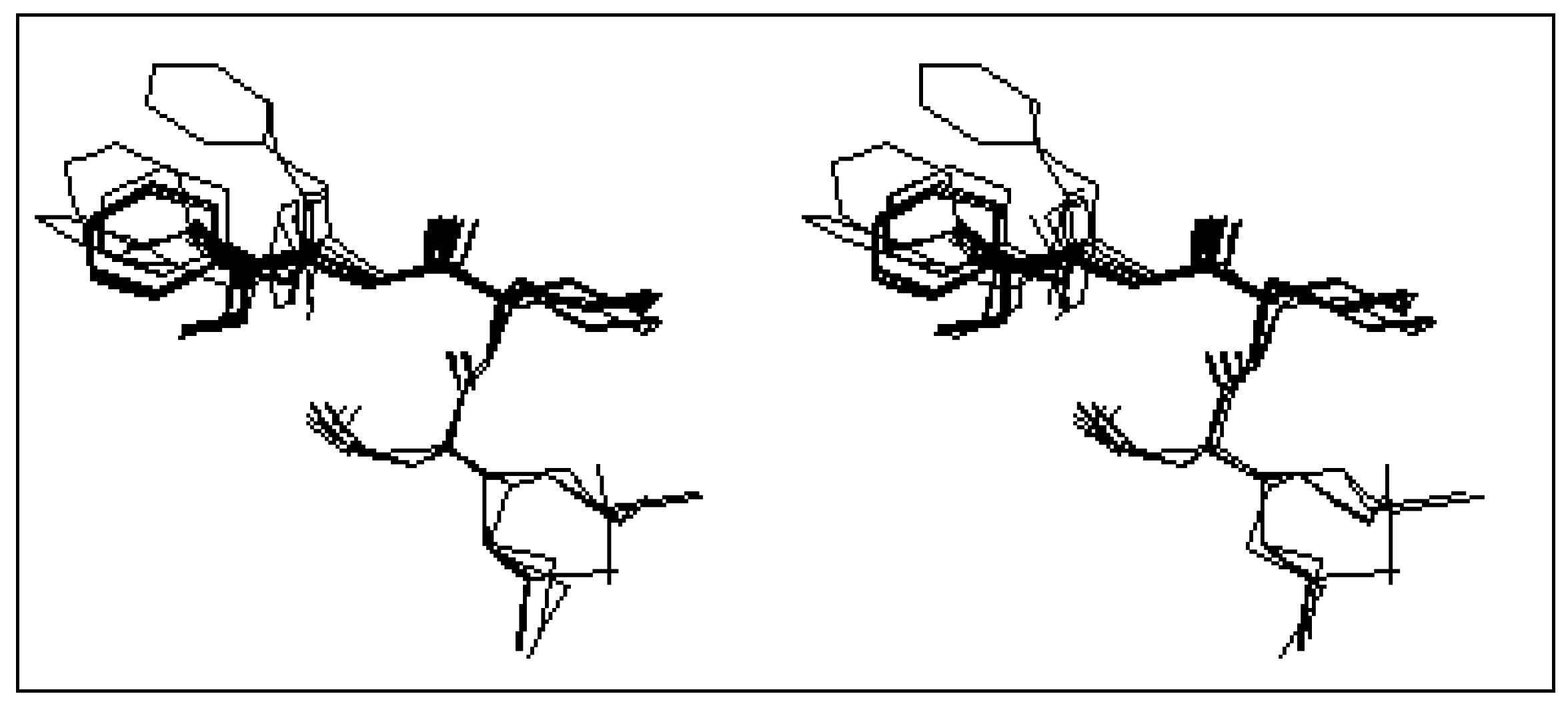
Concerning the constrained tripeptide formyl-Met-Acc6-Phe-OMe, the conformational analysis of the most twenty stable conformers (
table 2) shows a great tendency toward the β turn structure. Indeed, 17 conformers over the 20 most stable conformers, represent this structure. The stereoscopic superposition view of these conformers is given in
Figure 3.
Table 2.
Conformational characteristics of formyl-Met-Acc6-Phe-OMe a.
Table 2.
Conformational characteristics of formyl-Met-Acc6-Phe-OMe a.
| Conf. | Relative free energy ΔG b (kcal/mol) | Relative conformational energy ΔE c (kcal/mol) | Formyl | Met | Acc6 | Phe |
|---|
| 1 | 0.00 | 0.00 | CO | | | NH |
| 2 | 0.82 | 0.99 | CO | | | NH |
| 3 | 1.02 | 0.43 | CO | | | NH |
| 4 | 1.52 | 1.27 | CO | | | NH |
| 5 | 1.56 | 1.82 | | | | |
| 6 | 1.63 | 1.02 | CO | | NH | |
| 7 | 1.69 | 1.73 | CO | | | NH |
| 8 | 1.70 | 1.55 | CO | | | NH |
| 9 | 1.73 | 1.21 | CO | | | NH |
| 10 | 1.78 | 1.61 | CO | | | NH |
| 11 | 1.80 | 2.41 | CO | | | NH |
| 12 | 1.81 | 2.41 | CO | | | NH |
| 13 | 1.86 | 0.82 | CO | | | NH |
| 14 | 1.87 | 2.07 | CO | | | NH |
| 15 | 1.89 | 2.08 | CO | | | NH |
| 16 | 1.93 | 2.02 | CO | | | NH |
| 17 | 1.95 | 0.98 | | | | |
| 18 | 1.96 | 2.03 | CO | | | NH |
| 19 | 2.02 | 1.99 | CO | | | NH |
| 20 | 2.06 | 0.97 | CO | | | NH |
Figure 3.
Stereoscopic superposition view of the 17 conformers of formyl-Met-Acc6-Phe-OMe in β turn structure.
Figure 3.
Stereoscopic superposition view of the 17 conformers of formyl-Met-Acc6-Phe-OMe in β turn structure.
Table 3.
List of torsional angles for formyl-Met-Leu-Phe-OMe.
Table 3.
List of torsional angles for formyl-Met-Leu-Phe-OMe.
![Ijms 02 00001 i001]() |
Table 4.
List of torsional angles for formyl-Met-Acc6-Phe-OMe.
Table 4.
List of torsional angles for formyl-Met-Acc6-Phe-OMe.
![Ijms 02 00001 i002]() |
From these results, it appears clear that the parent peptide formyl-Met-Leu-Phe-OMe can adopt several types of structures in such way that we can not favor a precise structure, as compared to other structures. Therefore, it is not easy to extract the conformational characteristics of the chemotactic peptides using only the parent peptide formyl-Met-Leu-Phe-OMe.
The conformational analysis results of the geometrically constrained peptides formyl-Met-Acc6-Phe-OMe in which the α,α-disubstituted amino acid Acc6 gives it a certain rigidity, shows the preference of this tripeptide to adopt β turn conformation.
Taking into account the findings above, we can propose that the active structure of chemotactic peptide is the β turn structure preferred by the geometrically constrained peptide formyl-Met-Acc6-Phe-OMe. However, in the case of the parent peptide formyl-Met-Leu-Phe-OMe, we can suppose that its activity is due to its flexibility. This flexibility allows the molecule to fit the convenient structure (β turn) during the interaction with the receptor. This result is in perfect agreement with the "Zipper" model of Burgen [
17].
Finally, the comparison of results in
table 1 and
table 2 enables us to reject the proposal that the formyl group must be free of any intramolecular hydrogen bond in order to be available for the formation of the complex with the receptor. Indeed, among the 20 most stable structures of formyl-Met-Acc6-Phe-OMe, 18 conformers have the formyl group implicated in intramolecular hydrogen bonds, even though this peptide is six times more active than the parent peptide formyl-Met-Leu-Phe-OMe.


{kind=link}
{kind=link}
{kind=link}