STAT3, a Hub Protein of Cellular Signaling Pathways, Is Triggered by β-Hexaclorocyclohexane

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
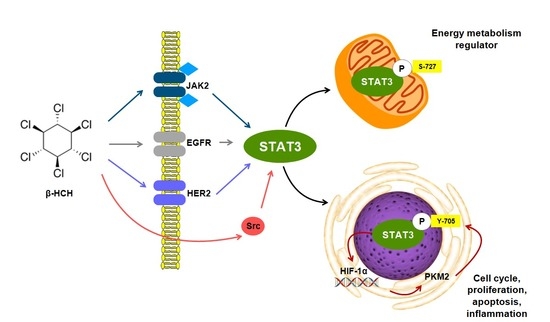
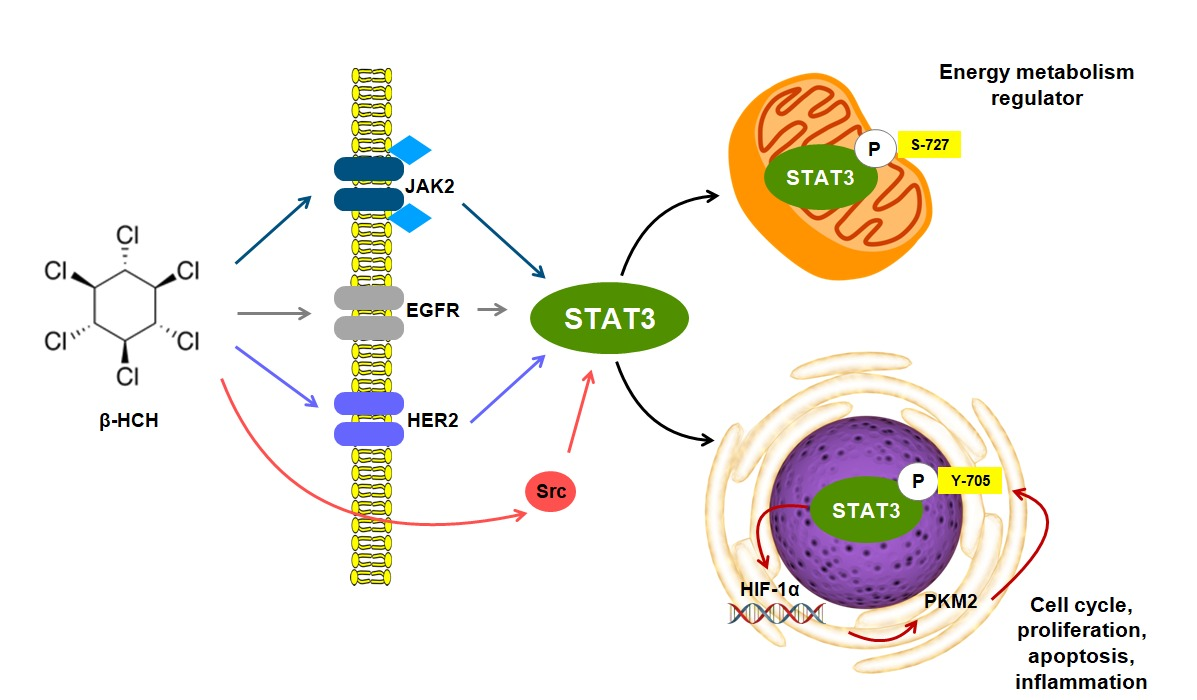{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
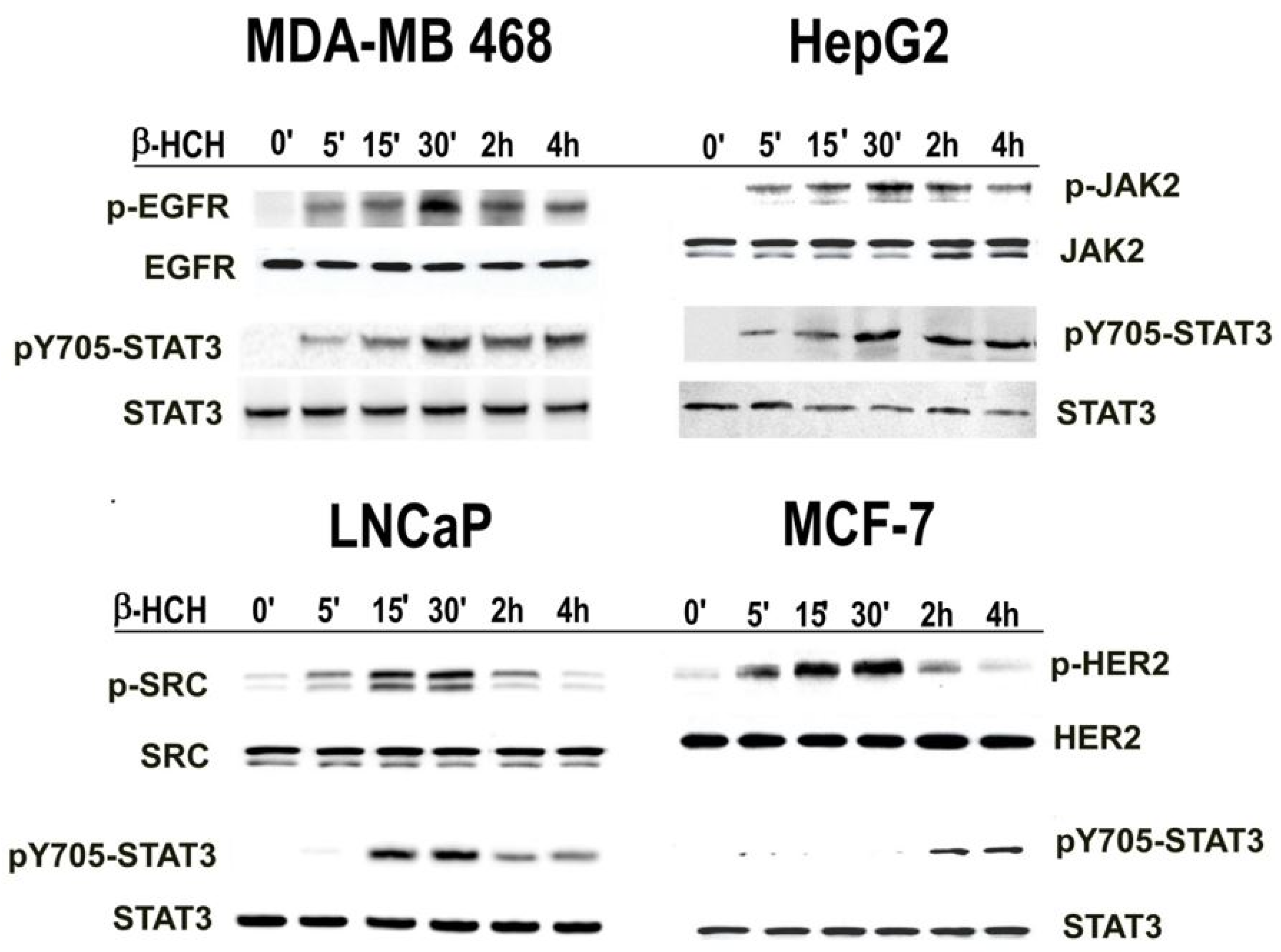
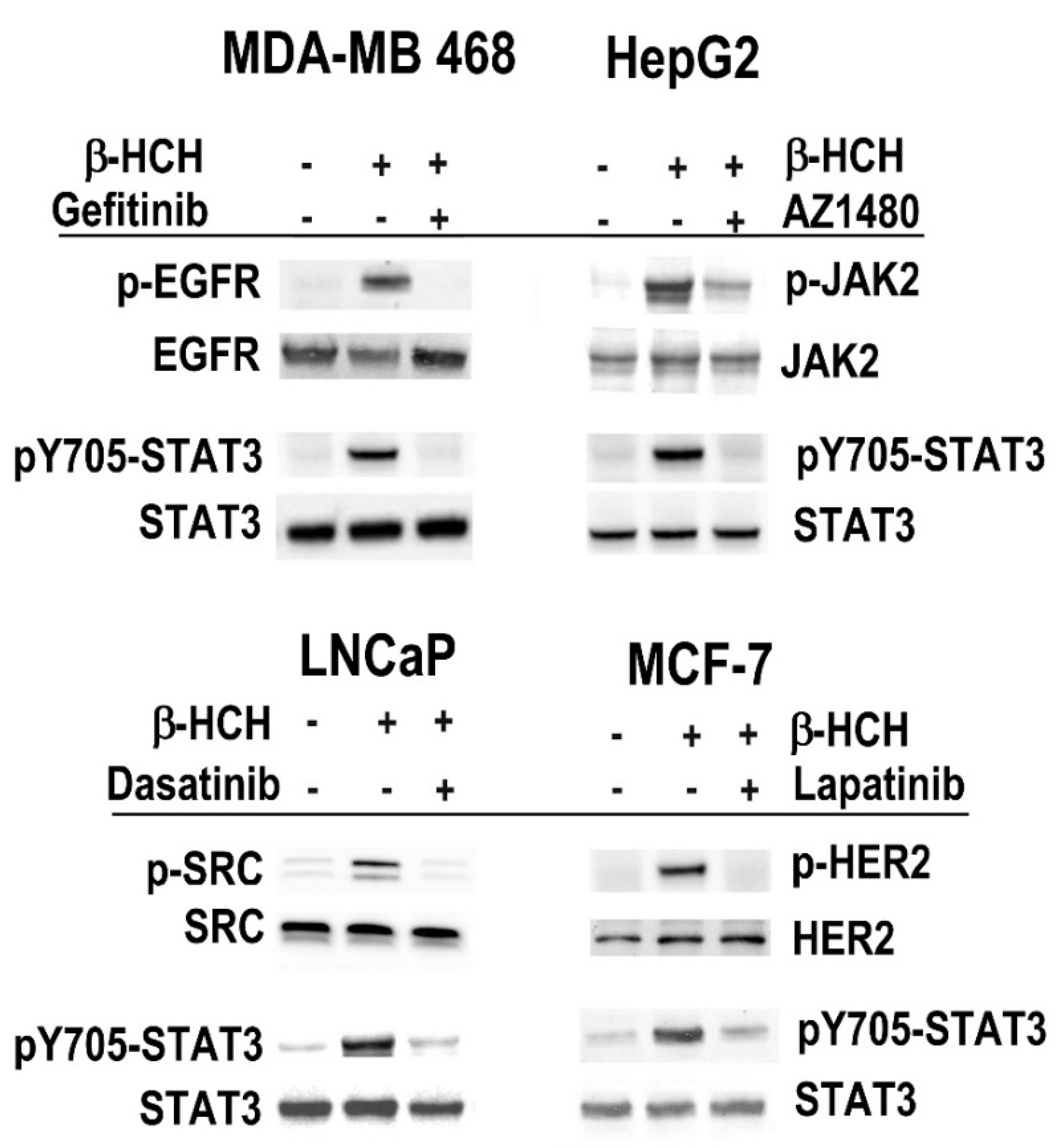
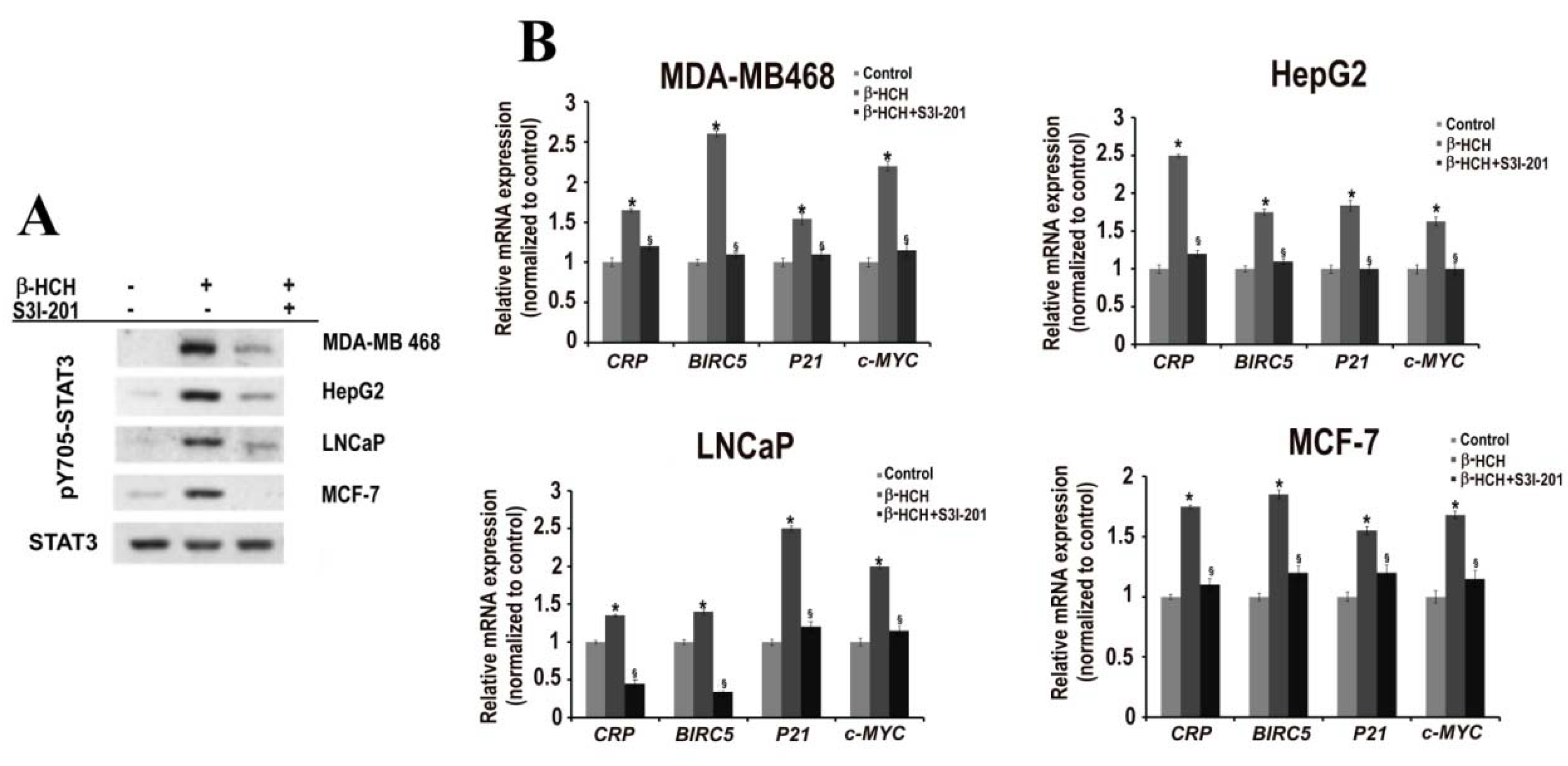
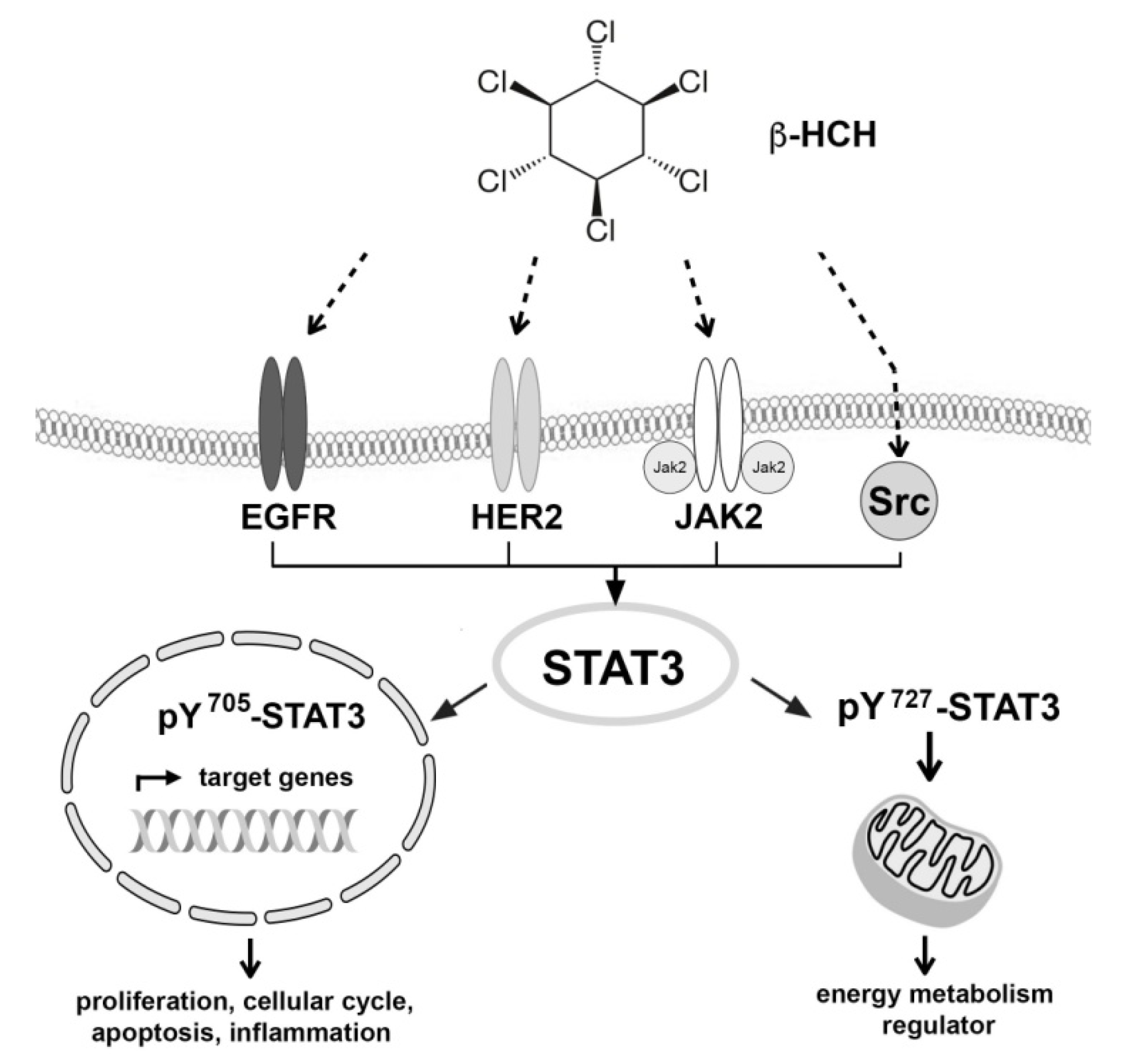
2.1. β-HCH Triggers Key Molecular Pathways in Different Cancer Cells
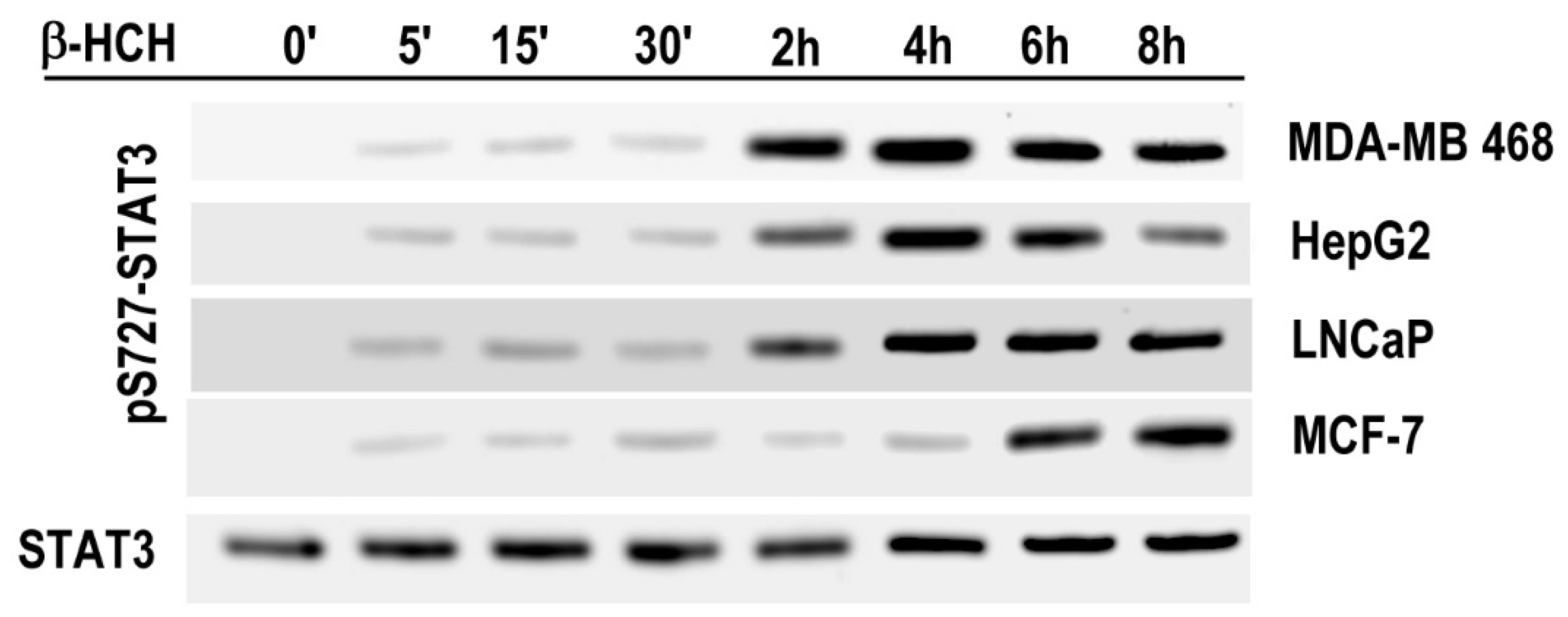
2.2. STAT3: Signal Transducer and Transcription Factor Mediating the Activity of β-HCH
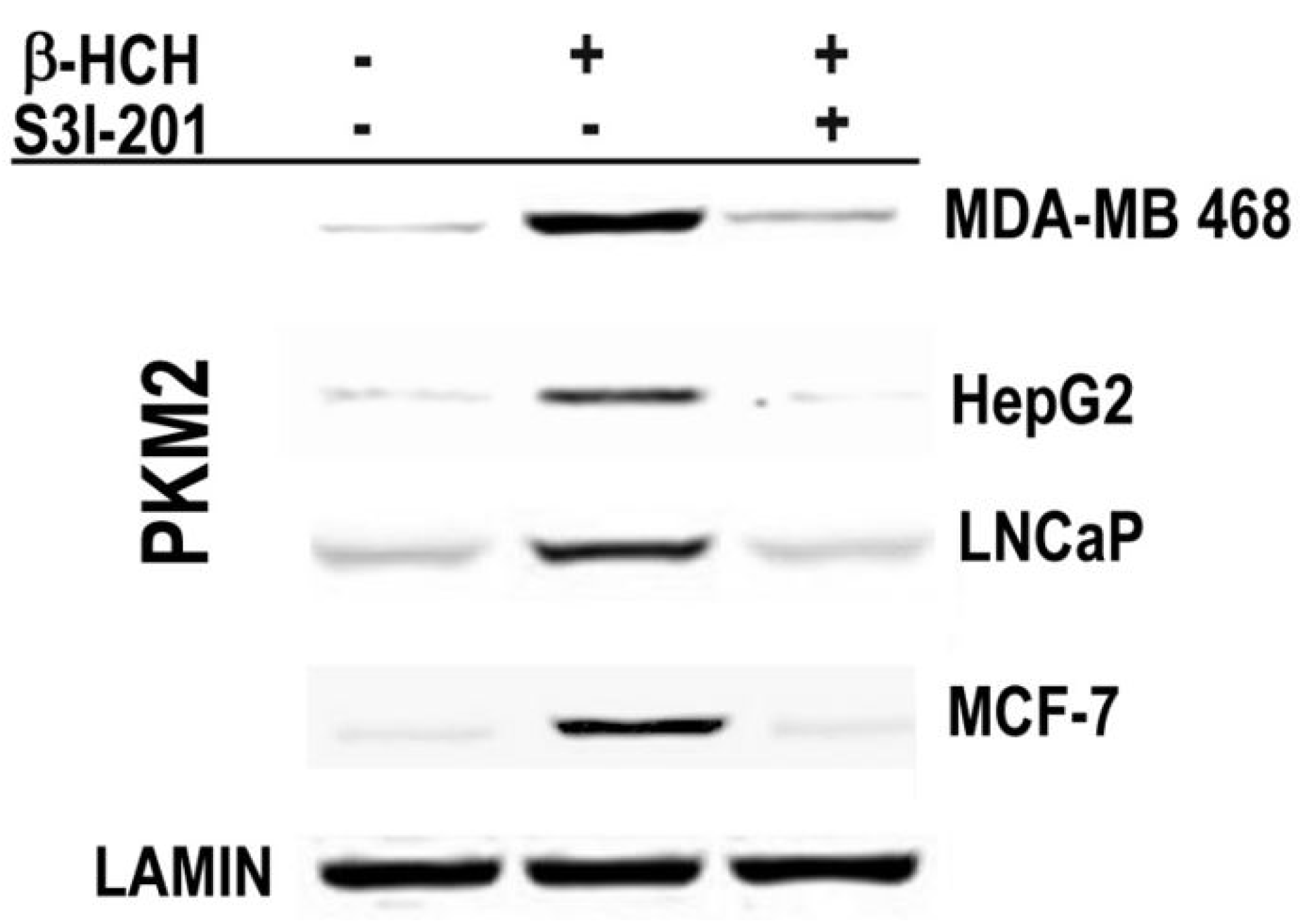
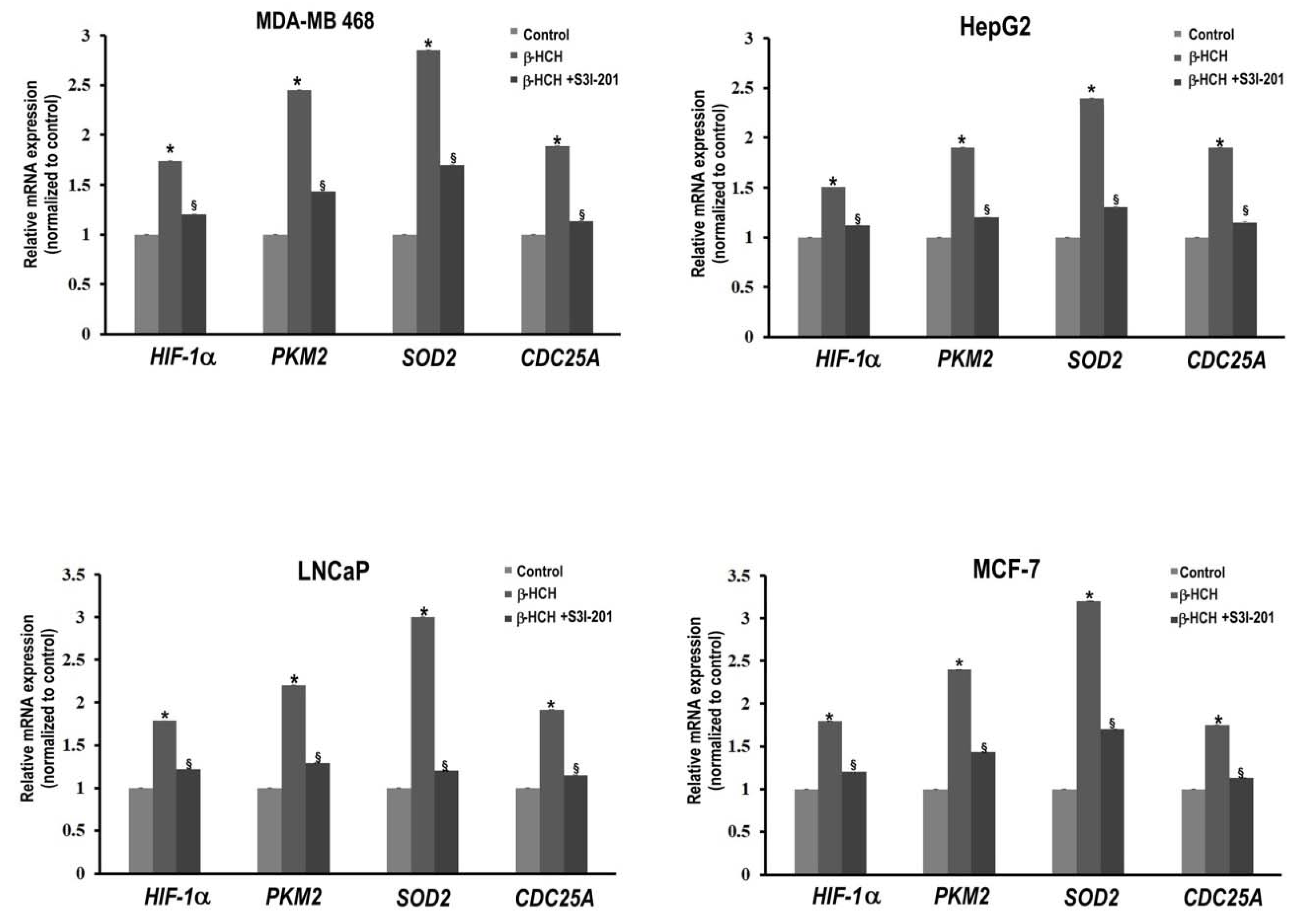
2.3. STAT3 as a Master Regulator of the Cell Metabolism
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Proteins Extraction and Immunoblotting
4.3. Extraction of RNA and RT-qPCR
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OCPs | Organochlorine pesticides |
| β-HCH | β-hexaclorocyclohexane |
| POPs | Persistent Organic Pollutants |
| EDCs | endocrine disrupting chemicals |
References
- Annamalai, J.; Namasivayam, V. Endocrine disrupting chemicals in the atmosphere: Their effects on humans and wildlife. Environ Int. 2015, 76, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Bonefeld-Jørgensen, E.C.; Ghisari, M.; Wielsøe, M.; Bjerregaard-Olesen, C.; Kjeldsen, L.S.; Long, M. Biomonitoring and hormone-disrupting effect biomarkers of persistent organic pollutants in vitro and ex vivo. Basic Clin. Pharmacol. Toxicol. 2014, 115, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Fagnocchi, L.; Poli, V.; Zippo, A. Enhancer reprogramming in tumor progression: A new route towards cancer cell plasticity. Cell. Mol. Life Sci. 2018, 75, 2537–2555. [Google Scholar] [CrossRef] [PubMed]
- Yang, M. A current global view of environmental and occupational cancers. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2011, 29, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Khatami, M.; Baglole, C.J.; Sun, J.; Harris, S.A.; Moon, E.Y.; Al-Mulla, F.; Al-Temaimi, R.; Brown, D.G.; Colacci, A.; et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 2015, 36, 36. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.; Emond, C.; Kim, M.J.; Antignac, J.P.; Le Bizec, B.; Clément, K.; Birnbaum, L.S.; Barouki, R. Toxicological function of adipose tissue: Focus on persistent organic pollutants. Environ. Health Perspect. 2013, 121, 162–169. [Google Scholar] [PubMed]
- Kim, M.J.; Pelloux, V.; Guyot, E.; Tordjman, J.; Bui, L.C.; Chevallier, A.; Forest, C.; Benelli, C.; Clément, K.; Barouki, R. Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ. Health Perspect. 2012, 120, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jain, M.; Mathur, V.; Feroz, S.M. Chronic Inflammation and Cancer: Paradigm on Tumor Progression, Metastasis and Therapeutic Intervention. Gulf J. Oncol. 2016, 1, 86–93. [Google Scholar]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lv, J.; Han, M.; Yang, Z.; Li, T.; Jiang, S.; Yang, Y. STAT3: The art of multi-tasking of metabolic and immune functions in obesity. Prog. Lipid Res. 2018, 70, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Camporeale, A.; Demaria, M.; Monteleone, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P.; Poli, V. STAT3 Activities and Energy Metabolism: Dangerous Liaisons. Cancers 2014, 6, 1579–1596. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α: The Warburg’s vicious circle. JAKSTAT 2012, 1, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.S.; Escada-Rebelo, S.; Correia, M.; Mota, P.C.; Ramalho-Santos, J. The non-genomic effects of endocrine-disrupting chemicals on mammalian sperm. Reproduction 2016, 151, R1–R13. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Kabil, A.; Kortenkamp, A. Cross-talk between non-genomic and genomic signalling pathways: Distinct effect profiles of environmental estrogens. Toxicol. Appl. Pharmacol. 2010, 245, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Loomis, D.; Guyton, K.; Grosse, Y.; El Ghissasi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K. International Agency for Research on Cancer Monograph Working Group, IARC, Lyon, France. Carcinogenicity of lindane, DDT, and 2,4-dichlorophenoxyacetic acid. Lancet Oncol. 2015, 16, 891–892. [Google Scholar] [CrossRef]
- Vijgen, J.; Yi, L.F.; Forter, M.; Lal, R.; Weber, R. The legacy of lindane and technical HCH production. Organohalog. Compd. 2006, 68, 899–904. [Google Scholar]
- Weber, R.; Gaus, C.; Tysklind, M.; Johnston, P.; Forter, M.; Hollert, H.; Heinisch, E.; Holoubek, I.; Lloyd-Smith, M.; Masunaga, S.; et al. Dioxin- and POP-contaminated sites, contemporary and future relevance and challenges: overview on background, aims and scope of the series. Environ. Sci. Pollut. Res. Int. 2008, 15, 363–393. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Porta, D.; Fantini, F.; De Felip, E.; Blasetti, F.; Abballe, A.; Dell’Orco, V.; Fano, V.; Ingelido, A.M.; Narduzzi, S.; Forastiere, F. A biomonitoring study on blood levels of beta-hexachlorocyclohexane among people living close to an industrial area. Environ. Health 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Narduzzi, S.; Porta, D.; Fantini, F.; Blasetti, F.; Davoli, M.; Forastiere, F. Sorveglianza Sanitaria ed Epidemiologica Della Popolazione Residente in Prossimità del Fiume Sacco, Rapporto Tecnico Attività 2013–2015; Dipartimento di Epidemiologia del Servizio Sanitario Regionale-Regione Lazio: Roma, Italy, 2016. [Google Scholar]
- Sharma, H.; Zhang, P.; Barber, D.S.; Liu, B. Organochlorine pesticides dieldrin and lindane induce cooperative toxicity in dopaminergic neurons: Role of oxidative stress. Neurotoxicology 2010, 31, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Briz, V.; Molina-Molina, J.M.; Sánchez-Redondo, S.; Fernández, M.F.; Grimalt, J.O.; Olea, N.; Rodríguez-Farré, E.; Suñol, C. Differential estrogenic effects of the persistent organochlorine pesticides dieldrin, endosulfan, and lindane in primary neuronal cultures. Toxicol. Sci. 2011, 120, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.; Scholze, M.; Kortenkamp, A. Mixtures of four organochlorines enhance human breast cancer cell proliferation. Environ. Health Perspect. 2001, 109, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 Inhibitor, AZD1480, Potently Blocks Stat3 Signaling and Oncogenesis in Solid Tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; González-González, M.E.; Barrera, D.; Díaz, L.; García-Becerra, R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: Preclinical and clinical evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar] [PubMed]
- Mukherjee, A.; Dhadda, A.S.; Shehata, M.; Chan, S. Lapatinib: A tyrosine kinase inhibitor with a clinical role in breast cancer. Expert Opin. Pharmacother. 2007, 8, 2189–2204, Review; Erratum in Expert Opin. Pharmacother. 2007, 8, 3085–3086. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Michelle, A.B.; Benjamin, G.; Harshani, R.L.; Richard Yip, M.L.; Richard, J.; Mark, M.M.L.; Nicholas, J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Monica Lind, P.; Salihovic, S.; Van Bavel, B.; Lind, L.; Ingelsson, E. Influence of persistent organic pollutants on oxidative stress in population-based samples. Chemosphere 2014, 114, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask STAT3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Kim, G.S.; Narasimhan, P.; Song, Y.S.; Chan, P.H. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J. Neurosci. 2009, 29, 7003–7014. [Google Scholar] [CrossRef] [PubMed]
- Barrè, A.; Vigneron, A.; Coqueret, A. The STAT3 transcription factor is a target for the Myc and retinoblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005, 280, 15673–15681. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Zhang, Y.; Yan, X.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, O.; Kim, H.L.; Weon, J.I.; Seo, Y.R. Endocrine-disrupting Chemicals: Review of Toxicological Mechanisms Using Molecular Pathway Analysis. J. Cancer Prev. 2015, 20, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Tsygankov, V.Y.; Boyarova, M.D.; Kiku, P.F.; Yarygina, M.V. Hexachlorocyclohexane (HCH) in human blood in the south of the Russian Far East. Environ. Sci. Pollut. Res. Int. 2015, 22, 14379–14382. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, C.; Lankadasari, M.B.; Aranjani, J.M.; Harikumar, K.B. Targeting oncogenic transcription factors by polyphenols: A novel approach for cancer therapy. Pharmacol. Res. 2018, 130, 273–291. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubini, E.; Altieri, F.; Chichiarelli, S.; Giamogante, F.; Carissimi, S.; Paglia, G.; Macone, A.; Eufemi, M. STAT3, a Hub Protein of Cellular Signaling Pathways, Is Triggered by β-Hexaclorocyclohexane. Int. J. Mol. Sci. 2018, 19, 2108. https://doi.org/10.3390/ijms19072108
Rubini E, Altieri F, Chichiarelli S, Giamogante F, Carissimi S, Paglia G, Macone A, Eufemi M. STAT3, a Hub Protein of Cellular Signaling Pathways, Is Triggered by β-Hexaclorocyclohexane. International Journal of Molecular Sciences. 2018; 19(7):2108. https://doi.org/10.3390/ijms19072108
Chicago/Turabian StyleRubini, Elisabetta, Fabio Altieri, Silvia Chichiarelli, Flavia Giamogante, Stefania Carissimi, Giuliano Paglia, Alberto Macone, and Margherita Eufemi. 2018. "STAT3, a Hub Protein of Cellular Signaling Pathways, Is Triggered by β-Hexaclorocyclohexane" International Journal of Molecular Sciences 19, no. 7: 2108. https://doi.org/10.3390/ijms19072108
APA StyleRubini, E., Altieri, F., Chichiarelli, S., Giamogante, F., Carissimi, S., Paglia, G., Macone, A., & Eufemi, M. (2018). STAT3, a Hub Protein of Cellular Signaling Pathways, Is Triggered by β-Hexaclorocyclohexane. International Journal of Molecular Sciences, 19(7), 2108. https://doi.org/10.3390/ijms19072108