A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update


Abstract
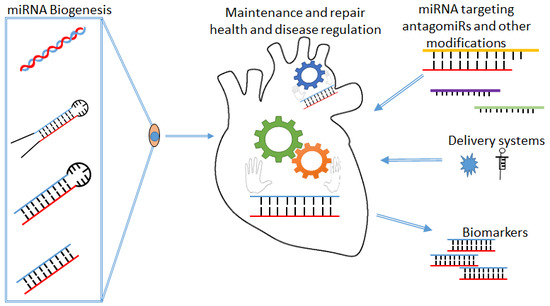
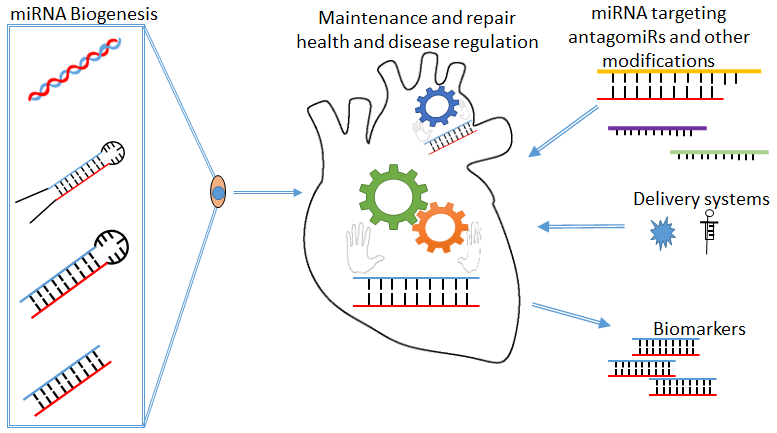
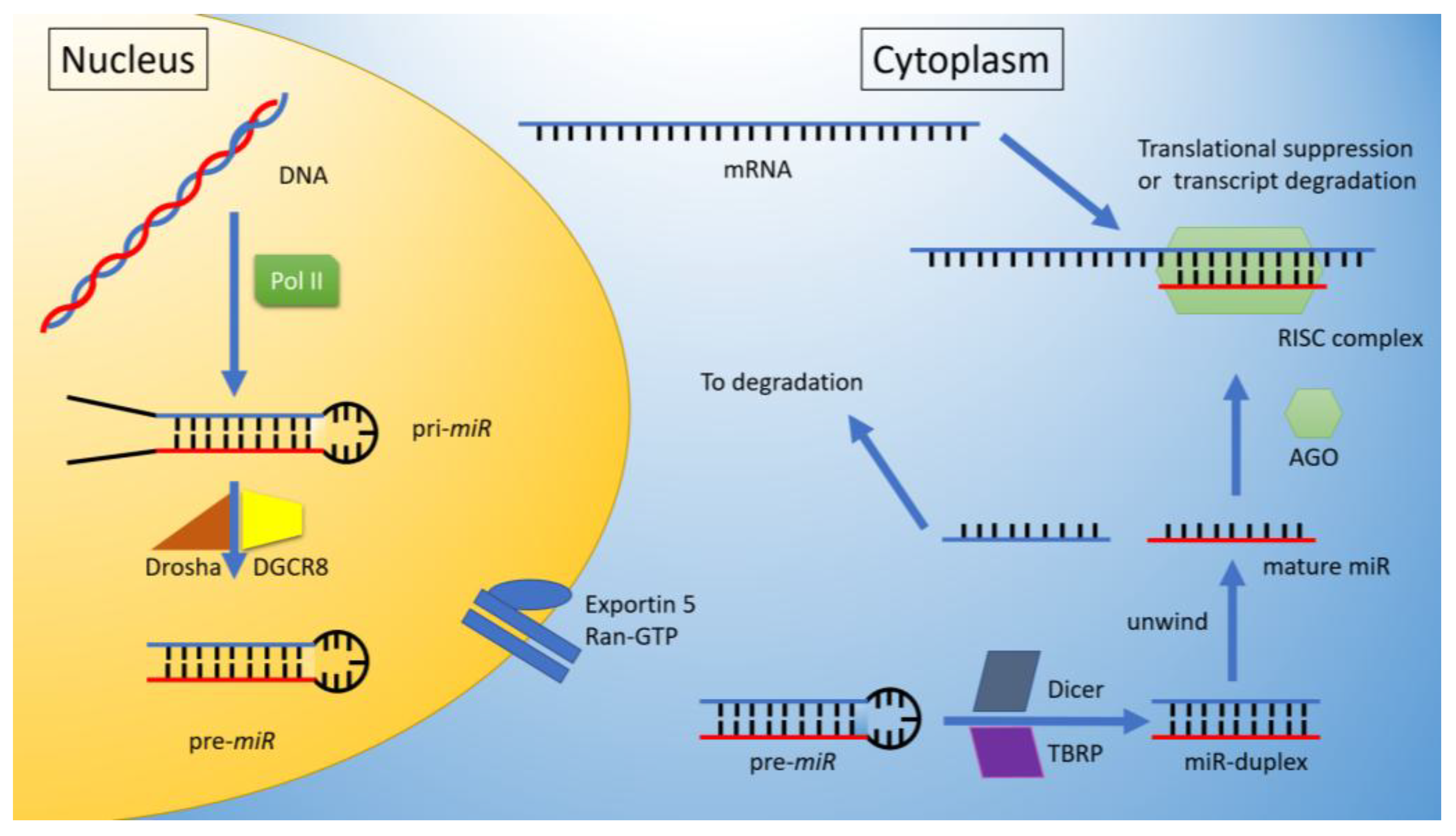
1. MicroRNA’s
miRNA Biogenesis
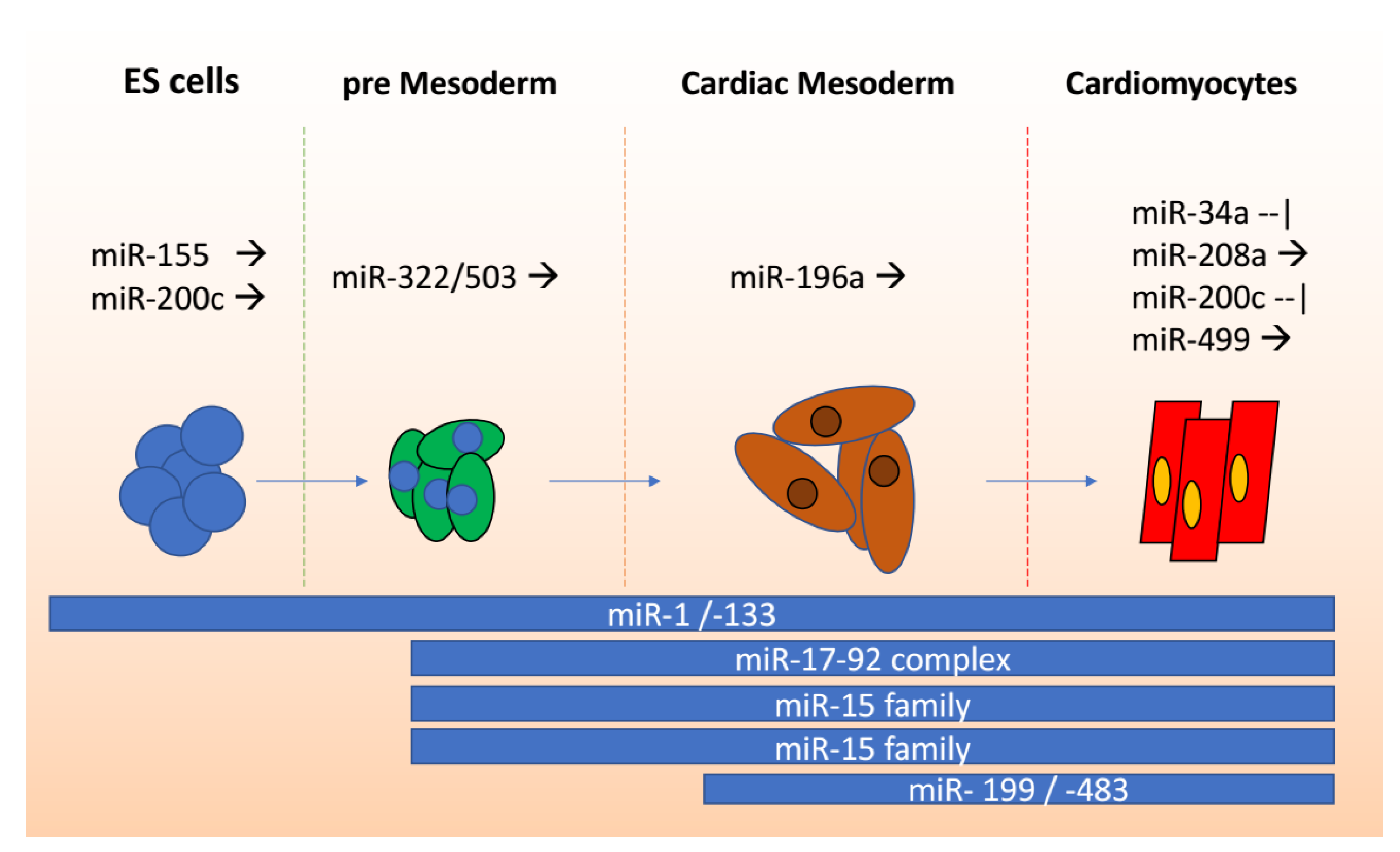
2. miRNAs in Cardiomyocytes
miRNAs in Stem Cells to Cardiomyocyte Differentiation
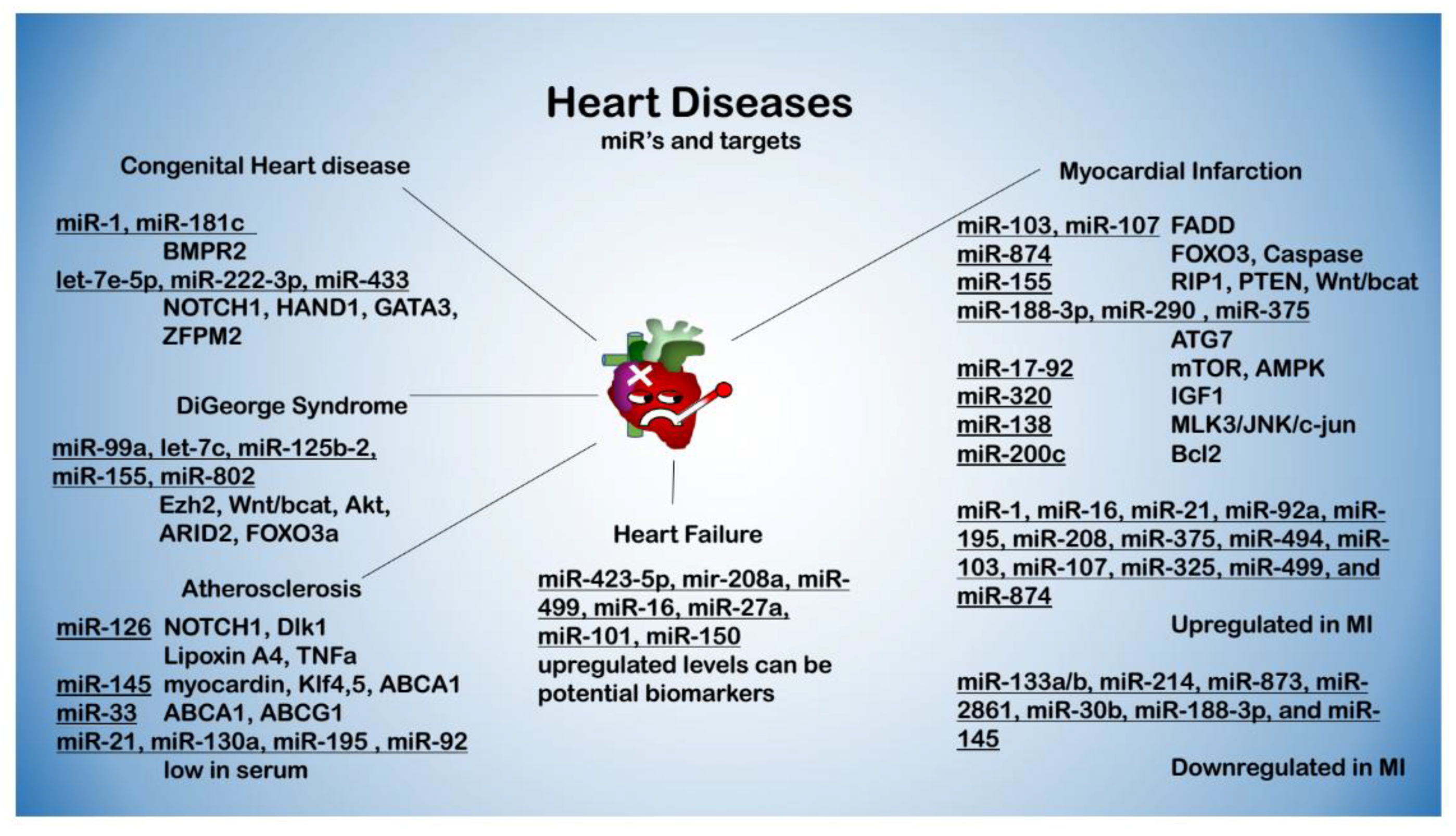
3. miRNAs in Cardiovascular Diseases
4. Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hydbring, P.; Badalian-Very, G. Clinical applications of microRNAs. F1000Research 2014, 2, 1–16. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Zhang, L.; Hao, C.; Li, J.; Qu, Y.; Bao, L.; Li, Y.; Yue, Z.; Zhang, M.; Yu, X.; Chen, H.; et al. Bioinformatics methods for identifying differentially expressed genes and signaling pathways in nano-silica stimulated macrophages. Tumour Biol. 2017, 6, 1010428317709284. [Google Scholar] [CrossRef] [PubMed]
- Romaine, S.P.R.; Tomaszewski, M.; Condorelli, G.; Samani, N. MicroRNAs in cardiovascular disease: An introduction for clinicians. Heart 2015, 101, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kowdley, K.V. MicroRNAs in Common Human Diseases. Genom. Proteom. Bioinform. 2012, 10, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Frost, R.J.A.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.L.T.; Schmittgen, T.D.; et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.-T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwisee processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting MicroRNAs in Cancer: Rationale, Strategies and Challenges. Nat. Rev. Drug Discov. 2013, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Demongeot, J.; Glade, N.; Moreira, A.; Vial, L. RNA relics and origin of life. Int. J. Mol. Sci. 2009, 10, 3420–3441. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Rajakaruma, C.; Caputo, M.; Emanueli, C. MicroRNAs in congenital heart disease. Ann. Transl. Med. 2015, 3, 333. [Google Scholar] [CrossRef] [PubMed]
- Alhendi, A.M.N.; Haider, S.; Jagannathan, S.; Anaissie, E.; Driscoll, J.J. MicroRNA theragnostics for the clinical management of multiple myeloma. Leukemia 2014, 28, 732–738. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-D.; Rushing, S.N.; Lieu, D.K.; Chan, C.W.; Kong, C.; Wilson, K.D.; Chiamvimonvat, N.; Boheler, K.R.; Wu, J.C.; Hajjar, R.J.; et al. Chiamvimonvat N, Li RA. Na+/Ca2+ exchanger is a determinant of excitation-contraction coupling in human embryonic stem cell-derived ventricular cardiomyocytes. Stem Cells Dev. 2010, 19, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.; Belaguli, N.; Flu, M.; Rowan, B.G.; Wei, L.; Weigel, N.L.; Booth, F.W.; Epstein, H.F.; Schwartz, R.J.; Balasubramanyam, A. Novel Phosphorylation Target in the Serum Response Factor MADS Box Regulates. Biochemistry 2003, 42, 7477–7486. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tao, Y.; Yu, W.; Schwartz, R.J. Brief report: Srf-dependent MiR-210 silences the sonic hedgehog signaling during cardiopoesis. Stem Cells 2013, 31, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schwartz, R.J. Transient Mesp1 expression: A driver of cardiac cell fate determination. Transcription 2013, 4, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, E.; Mallat, Y.; Lefebvre, F.; Diguet, N.; Escoubet, B.; Blanc, J.; De Windt, L.J.; Catalucci, D.; Vandecasteele, G.; Li, Z.; et al. An SRF/miR-1 axis regulates NCX1 and Annexin A5 protein levels in the normal and failing heart. Cardiovasc. Res. 2013, 98, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; An, X.; Niu, L. Role of microRNAs in cardiac development and disease. Exp. Ther. Med. 2017, 13, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chaitra, K.L.; Ulaganathan, K.; James, A.; Ananthapur, V.; Nallari, P. miRNA regulation during cardiac development and remodeling in cardiomyopathy. EXCLI J. 2013, 12, 980–992. [Google Scholar] [PubMed]
- Wu, K.H.; Xiao, Q.R.; Yang, Y.; Xu, J.L.; Zhang, F.; Liu, C.M.; Zhang, Z.M.; Lu, Y.Q.; Huang, N.P. MicroRNA-34a modulates the Notch signaling pathway in mice with congenital heart disease and its role in heart development. J. Mol. Cell. Cardiol. 2018, 114, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Stahlhut, C.; Giraldez, A.J. miR-1-2 Gets to the Heart of the Matter. Cell 2007, 129, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, X.; Wang, Z.; Wu, J. MicroRNA-1 in cardiac diseases and cancers. Korean J. Physiol. Pharmacol. 2014, 18, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Len, H.; Shi, X.; Ji, J.; Fu, J.; Len, H. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed. Pharmacother. 2017, 90, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Jiao, K. Functions of miRNAs during mammalian heart development. Int. J. Mol. Sci. 2016, 17, 789. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.; Park, D.; Rotllan, N.; Chamorro-Jorganes, A.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.; Hernando, E. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Hee, Y.S.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.; Jason, E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Harold, S.; et al. MicroRNA Regulation of Cell Lineages in Mouse and Human Embryonic Stem Cells. Cell Stem Cell. 2009, 2, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Garcia-Gras, E.; Wycuff, D.R.; Marriot, S.J.; Kadeer, N.; Yu, W.; Olson, E.N.; Garry, D.J.; Parmacek, M.S.; Schwartz, R.J. Identification of direct serum-response factor gene targets during Me2SO-induced P19 cardiac cell differentiation. J. Biol. Chem. 2005, 280, 19115–19126. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.; Hao, B.; Guan, D.; Li, M.; Lu, J.; Yang, Y.; Wu, B.; Wu, S.; Webb, S.; Liang, Y.; et al. Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovasc. Res. 2018, 114. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Soibam, B.; Benham, A.; Xu, X.; Chopra, M.; Peng, X.; Yu, W.; Bao, W.; Liang, R.; Azares, A.; et al. miR-322/-503 cluster is expressed in the earliest cardiac progenitor cells and drives cardiomyocyte specification. Proc. Natl. Acad. Sci. USA 2016, 113, 9551–9556. [Google Scholar] [CrossRef] [PubMed]
- Lieu, D.K.; Fu, J.; Chiamvimonvat, N.; Tung, K.W.C.; McNerney, G.P.; Huser, T.; Keller, G.; Kong, C.-W.; Li, R.A. Mechanism-Based Facilitated Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Arrhythm. Electrophysiol. 2013, 6, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, N.; Chellappan, M.; Adler, A.F.; Kirkton, R.D.; Wu, T.; Addis, R.C.; Bursac, N.; Leong, K.W. Transcription Factors MYOCD, SRF, Mesp1 and SMARCD3 Enhance the Cardio-Inducing Effect of GATA4, TBX5, and MEF2C during Direct Cellular Reprogramming. PLoS ONE 2013, 8, e63577. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.; Egemnazarov, B.; Finch, E.; Zhan, L.; Payne, A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V. MicroRNA-mediated in vitro and in vivo Direct Reprogramming of Cardiac Fibroblasts to Cardiomyocytes. Circ. Res. 2013, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-M. Creation of engineered cardiac tissue in vitro from mouse embryonic stem cells. Circulation 2006, 113, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Ieda, M. Critical Factors for Cardiac Reprogramming. Circ. Res. 2013, 111, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Belian, E.; Noseda, M.; Abreu Paiva, M.S.; Leja, T.; Sampson, R.; Schneider, M.D. Forward Programming of Cardiac Stem Cells by Homogeneous Transduction with MYOCD plus TBX5. PLoS ONE 2015, 10, e0125384. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Terry, S.; Gyorke, S. Mir-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56α and causing camkii-dependent hyperphosphorylation of RyR2. Circ. Res. 2015, 104, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.S.; Palanisamy, K.; Chang, S.S.; Sun, K.T.; Chen, K.B.; Li, P.C.; Lin, T.C.; Li, C.Y. Plasma exosomal miR-223 expression regulates inflammatory responses during cardiac surgery with cardiopulmonary bypass. Sci. Rep. 2017, 7, 10807. [Google Scholar] [CrossRef] [PubMed]
- Bondue, A.; Blanpain, C. MESP1. A Key Regulator of Cardiovascular Lineage Commitment. Circ. Res. 2010, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Islas, J.F.; Liu, Y.; Weng, K.-C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef] [PubMed]
- Joladarashi, D.; Thandavarayan, R.A.; Babu, S.S. Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. Int. J. Mol. Sci. 2014, 15, 15891–15911. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Xia, D.; Cheng, Z.; Zhang, G. Role of Sprouty1 (Spry1) in the pathogenesis of atrial fibrosis. Pathol. Res. Pract. 2018, 214, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gong, Y.; Tang, Y.; Li, H.; He, Q.; Gower, L.; Llaw, L.; Friesel, R. Spry1 and Spry4 Differentially Regulate Human Aortic Smooth Muscle Cell Phenotype via Akt/FoxO/Myocardin Signaling. PLoS ONE 2013, 8, e58746. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jaber, V.; Percy, M.E.; Lukiw, W.J. A microRNA cluster (let-7c, miRNA-99a, miRNA-125b, miRNA-155 and miRNA-802) encoded at chr21q21.1-chr21q21.3 and the phenotypic diversity of Down’s syndrome (DS; trisomy 21). J. Nat. Sci 2017, 3. [Google Scholar] [CrossRef]
- Li, Y.; Maegdefessel, L. My heart will go on—Beneficial effects of anti-MiR-30 after myocardial infarction. Ann. Transl. Med. 2016, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Tseliou, E.; de Couto, G.; Terrovitis, J.; Sun, B.; Weixin, L.; Marbán, L.; Marbán, E. Angiogenesis, cardiomyocyte proliferation and anti-fibrotic effects underlie structural preservation post-infarction by intramyocardially-injected cardiospheres. PLoS ONE 2014, 9, e88590. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef]
- Ling, X.; Yao, D.; Kang, L.; Zhou, J.; Zhou, Y.; Dong, H. Involment of RAS/ERK1/2 signaling and MEF2C in miR-155-3p inhibition-triggered cardiomyocyte differentiation of embryonic stem cell. Oncotarget 2017, 8, 84403–84416. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, E.; Gorniak, P.; Prusisz, W.; Kiliszek, P.; Szydlowski, M.; Sewastianik, T.; Bialopiotrowicz, E.; Polak, A.; Prochorec-Sobieszek, M.; Szumera-Cieckiewicz, A.; et al. MiR-155 Amplifies AKT and NFkB Signaling By Targeting Multiple Regulators of BCR Signal in DLBCL. Blood 2015, 126, 2455. [Google Scholar]
- Schulte, C.; Zeller, T. microRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc. Diagn. Ther. 2015, 5, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Bensemlali, M.; Bajolle, F.; Ladouceur, M.; Fermont, L.; Lévy, M.; Le Bidois, J.; Salomon, L.J.; Bonnet, D. Associated genetic syndromes and extracardiac malformations strongly influence outcomes of fetuses with congenital heart diseases. Arch. Cardiovasc. Dis. 2016, 109, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Heart disease and stroke statistics-2016 update a report from the American Heart Association. Circulation 2016, 133, e38–e48. [Google Scholar] [CrossRef] [PubMed]
- Mercola, M.; Ruiz-lozano, P.; Schneider, M.D. Cardiac muscle regeneration: Lessons from development Cardiac muscle regeneration: Lessons from development. Genes Dev. 2011, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Bigdelian, H.; Sedighi, M. The role of preoperative sildenafil therapy in controlling of postoperative pulmonary hypertension in children with ventricular septal defects. J. Cardiovasc. Thorac. Res. 2017, 9, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Rossetti, L.; Faggian, G.; Luciani, G.B. Long-Term Follow-Up Study of Temporary Tricuspid Valve Detachment as Approach to VSD Repair without Consequent Tricuspid Dysfunction. Tex. Heart Inst. J. 2016, 43, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, Y.; Ma, X.; Wang, H.; Zhang, J.; Luo, X.; Chen, W.; Wu, Y.; Meng, Y.; Zhang, J.; et al. Roles of miR-1-1 and miR-181c in ventricular septal defects.le. Int. J. Cardiol. 2013, 168, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c Regulates the Mitochondrial Genome, Bioenergetics, and Propensity for Heart Failure In Vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef] [PubMed]
- Landthaler, M.; Abdullah, Y.; Tuschi, T. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. 2004, 14, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Romito, A.; Borel, C.; Gehrig, C.; Gagnebin, M.; Falconnet, E.; Izzo, A.; Altucci, L.; Banfi, S.; Antonarakis, S.E.; et al. Cardiomyogenesis is controlled by the miR-99a/let-7c cluster and epigenetic modifications. Stem Cell Res. 2014, 12, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Chen, J.; Li, W.; Bao, C.; Fu, Q. Targeting miR-155 to Treat Experimental Scleroderma. Sci. Rep. 2016, 6, 20314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Li, X.; He, S.; Yao, J.; Wang, X.; Zhang, D.; Sun, X. MicroRNA-155 promotes tumor growth of human hepatocellular carcinoma by targeting ARID2. Int. J. Oncol. 2018, 48, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Gu, J.; Lei, Z.; Li, M.; Zhao, J.; Zhang, H.; Li, X. microRNA-155 regulates cell proliferation and invasion by targeting FOXO3a in glioma. Oncol. Rep. 2013, 30, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.P.M.; Fayad, Z.A.; Dweck, M.R. New methods to image unstable atherosclerotic plaques. Atherosclerosis 2018, 272, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Codagnone, M.; Recchiuti, A.; Lanuti, P.; Pierdomenico, A.M.; Cianci, E.; Patruno, S.; Mari, V.C.; Simiele, F.; Di Tomo, P.; Pandolfi, A.; et al. Lipoxin A4stimulates endothelial miR-126-5p expression and its transfer via microvesicles. FASEB J. 2017, 31, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Voora, D. The Last Line of Defense Against Atherosclerosis. Sci. Transl. Med. 2014, 6, 228ec51. [Google Scholar] [CrossRef]
- Boon, R.A.; Dimmeler, S. MicroRNA-126 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, e15–e16. [Google Scholar] [CrossRef] [PubMed]
- Faccini, J.; Ruidavets, J.B.; Cordelier, P.; Martins, F.; Maoret, J.J.; Bongard, V.; Ferrières, J.; Roncalli, J.; Elbaz, M.; Vindis, C. Circulating MIR-155, MIR-145 and let-7c as diagnostic biomarkers of the coronary artery disease. Sci. Rep. 2017, 7, 42916. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; Van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; Mcdaniel, A.L.; Marshall, M.; Van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and reduces VLDL triglycerides Katey. Nature 2012, 478, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Shiuchi, T.; Cui, T.-X.; Wu, L.; Nakagami, H.; Takeda-Matsubara, Y.; Iwai, M.; Horiuchi, M. ACE Inhibitor Improves Insulin Resistance in Diabetic Mouse Via Bradykinin and NO. Hypertension 2002, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.; Jin, L. miRNA-145 is associated with spontaneous hypertension by targeting SLC7A1. Exp. Ther. Med. 2017, 15, 48–552. [Google Scholar] [CrossRef]
- Wang, G.-K.; Zhu, J.-Q.; Zhang, J.-T.; Li, Q.; Li, Y.; He, J.; Qin, Y.-W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Thum, T. Non-coding RNAs in cardiac remodeling and heart failure. Circ. Res. 2013, 113, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Dong, Y.-H.; Du, W.; Shi, C.-Y.; Wang, K.; Tariq, M.-A.; Wang, J.-X.; Li, P.-F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Frazier, D.P.; Thompson, J.W.; Haliko, S.; Li, H.; Wasserlauf, B.J.; Spiga, M.G.; Bishopric, N.H.; Webster, K.A. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J. Exp. Biol. 2004, 207, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Li, Q.; Wang, K.; Wang, Y.; Jiao, J.; Feng, C.; Zhou, L.; Gong, Y.; Zhou, Z.; et al. MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circ. Res. 2015, 117, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, F.; Liu, C.; An, T.; Zhang, J.; Zhou, L.; Wang, M.; Dong, Y.; Li, N.; Gao, J.; et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016, 23, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.-Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Satoo, K.; Suzuki, H.; Fujioka, Y.; Ohsumi, O.; Inagaki, F.; Noda, N.N. Atg7 Activates an Autophagy-Essential Ubiquitin-like Protein Atg8 through Multi-Step Recognition. J. Mol. Biol. 2018, 430, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Sermersheim, M.A.; Park, K.H.; Gumpper, K.; Adesanya, T.M.A.; Song, K.; Tan, T.; Ren, X.; Yang, J.; Zhu, H.; Heart, D.; et al. MicroRNA regulation of autophagy in cardiovascular disease. Front. Biosci. (Landmark Ed.) 2017, 22, 48–65. [Google Scholar] [PubMed]
- He, X.; Li, C.; Ke, R.; Luo, L.; Huang, D. Down-regulation of adenosine monophosphate—Activated protein kinase activity: A driver of cancer. Tumor Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Samarel, A.M. IGF-1 overexpression rescues the failing heart. Circ. Res. 2002, 90, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; Van Der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Wan, L.; Zhao, D.; Qu, X.; Cai, F.; Huo, R.; Wang, N.; Zhu, J.; Zhang, C.; Zheng, F.; et al. Mild hypoxia-induced cardiomyocyte hypertrophy via up-regulation of HIF-1α-mediated TRPC signalling. J. Cell. Mol. Med. 2012, 16, 2022–2034. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.M.; Lee, Y.K.; Chan, Y.C.; Lai, W.H.; Fung, M.L.; Li, R.A.; Siu, C.W.; Tse, H.F. Exogenous expression of HIF-1?? promotes cardiac differentiation of embryonic stem cells. J. Mol. Cell. Cardiol. 2010, 48, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xue, H.; Jin, Q.-H.; Guo, J.; Chen, Y.-D. MiR-138 protects cardiac cells against hypoxia through modulation of glucose metabolism by targetting pyruvate dehydrogenase kinase 1. Biosci. Rep. 2017, 37, BSR20170296. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, S.; Guo, C.; Li, J.; Sang, W. Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. Int. J. Mol. Med. 2017, 39, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Roach, E.S.; Golomb, M.R.; Adams, R.; Biller, J.; Daniels, S.; Ferriero, D.; Jones, B.V.; Kirkham, F.J.; Scott, R.M.; Smith, E.R. Management of Stroke in Infants and Children a Scientific Statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke 2008, 39, 2644–2691. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Rawnsley, D.R.; Goddard, L.M.; Pan, W.; Cao, X.J.; Jakus, Z.; Zheng, H.; Yang, J.; Arthur, J.S.C.; Whitehead, K.J.; et al. The Cerebral Cavernous Malformation Pathway Controls Cardiac Development via Regulation of Endocardial MEKK3 Signaling and KLF Expression. Dev. Cell 2015, 32, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Wada, T.; Gramignoli, R.; Li, S.; Strom, S.C.; Huang, M.; Xie, W. MicroRNA hsa-miR-613 Targets the Human LXRα Gene and Mediates a Feedback Loop of LXRα Autoregulation. Mol. Endocrinol. 2011, 25, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, D.; Wu, Y.; Liu, X.; Zhu, J.; Tang, Y. MiR-155 inhibits proliferation and invasion by directly targeting PDCD4 in non-small cell lung cancer. Thorac. Cancer 2017, 8, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, Y.; Wang, C.; Qiu, D.; Zhou, K.; Hua, Y.; Li, Y. miRNAs as biomarkers for diagnosis of heart failure. Medicine 2017, 22, e6825. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y. MicroRNA and Heart Failure. Int. J. Mol. Sci. 2016, 17, 502. [Google Scholar] [CrossRef] [PubMed]
- Kwekkeboom, R.F.J.; Lei, Z.; Doevendans, P.A.; Musters, R.J.P.; Sluijter, J.P.G. Targeted delivery of miRNA therapeutics for cardiovascular diseases: Opportunities and challenges. Clin. Sci. 2014, 127, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.G.; Cavarretta, E. microRNAs in Cardiovascular Diseases Current Knowledge and the Road Ahead. J. Am. Coll. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Gomes da Silva, A.M.; Silbiger, V.N. miRNAs as biomarkers of atrial fibrillation. Biomarkers 2014, 19, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Bracken, C.P.; Szubert, J.M.; Goodall, G. On measuring miRNAs after transient transfection of mimics or antisense inhibitors. PLoS ONE 2013, 8, e55214. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Li, M.L.; Fang, Z.F.; Hu, X.Q.; Liu, Q.M.; Liu, Z.J.; Tang, L.; Zhao, Y.S.; Zhou, S. Overexpression of MicroRNA-1 improves the efficacy of mesenchymal stem cell transplantation after myocardial infarction. Cardiology 2013, 125, 18–30. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Regulation | Targets | References |
|---|---|---|---|
| miR-1/-133 | Proliferation and muscle growth | SRF, MEF2c, MyoD, Hand2, Myocardin | [23] |
| Signal mesoderm formation | Twist | [34] | |
| miR-1 | Conduction | NCX1 | [22] |
| Signaling | Repression of HDAC4 Activation of MEF2 | [34,35] | |
| miR-1-2 | Repolarization | Kcnd2 | [26,27] |
| miR-15 family | Cell Cycle | Repression of HSP-20 | [30] |
| miR-17-92 complex | Second heart field | BMP signaling, SMAD repression Isl1, Tbx1 | [30] |
| Signaling | Repression of PTEN | [9,29,30] | |
| Signaling | mTOR | [31] | |
| miR-34a | Specification | NOTCH1, Dlk1, Jagged, Hey, Hes | [24,25] |
| miR-155-3p | Regulates | MEF2c, KRAS (activate contractile factors) | [20,34,35] |
| miR-208 | Hypertrophy and muscle growth Myosin Heavy chain | a (208a), b (208b) | [32,33] |
| Conduction | GATA4, CX40 | ||
| miR-199/-483 | Signal mesoderm formation | Repression of Dlk-1 | [34] |
| miR-200c | Cardiac TF Conduction system | GATA4, TBX5, SRF CACNA1C, KCNJ2, SCN5A | [36] |
| miR322/503 | Mesoderm formation | Celf1 | [37] |
| miR-1/-499 | Electrical/conduction | Upregulates Kir2.1, Kv1.4, HERG, and DHPR Downregulates HCN4 | [18,38] |
| miR-1/-133/-208/-499 | Enhanced ES conversion | [33,37,39,40] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islas, J.F.; Moreno-Cuevas, J.E. A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update. Int. J. Mol. Sci. 2018, 19, 2075. https://doi.org/10.3390/ijms19072075
Islas JF, Moreno-Cuevas JE. A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update. International Journal of Molecular Sciences. 2018; 19(7):2075. https://doi.org/10.3390/ijms19072075
Chicago/Turabian StyleIslas, Jose Francisco, and Jorge Eugenio Moreno-Cuevas. 2018. "A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update" International Journal of Molecular Sciences 19, no. 7: 2075. https://doi.org/10.3390/ijms19072075
APA StyleIslas, J. F., & Moreno-Cuevas, J. E. (2018). A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update. International Journal of Molecular Sciences, 19(7), 2075. https://doi.org/10.3390/ijms19072075