Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology


Abstract
{kind=link}
1. Introduction
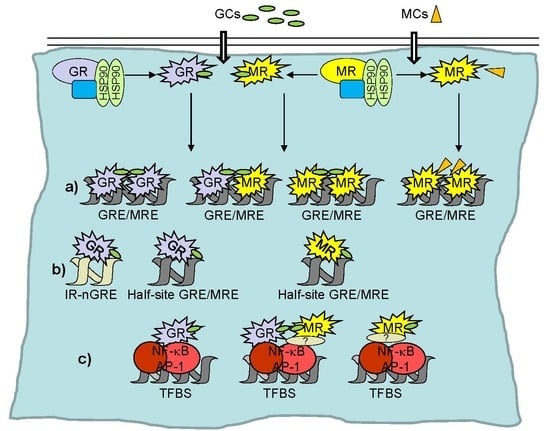
2. Transcriptional Regulation by GR and MR
3. Systemic and Cutaneous Glucocorticoid/Mineralocorticoid Production and Regulation
4. GC Signaling Exerts Crucial Roles in Skin Development
4.1. Development of the Epidermis and Its Appendages
4.2. Gain and Loss of Function Mouse Models for Studying GR and MR in Skin Development
5. GR and MR in Adult Skin Homeostasis
5.1. Expression and Function of GR and MR in Cultured Keratinocytes
5.2. Cutaneous Manifestations of Imbalances in GCs and Their Receptors
5.3. Strategies to Separate the Therapeutic and Adverse GC Effects
5.3.1. Skin Atrophy, Delayed Wound Healing, and Aging
5.3.2. Cutaneous Inflammation
6. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GCs | Glucocorticoids |
| MCs | Mineralocorticoids |
| GR | GC receptor |
| MR | MC receptor |
| GRE | GC/MC response element |
| HSD11B | 11 beta hydroxysteroid dehydrogenase |
| TF | Transcription factor |
| SC | Stratum corneum |
| GREKO | GR epidermal KO |
| MREKO | MR epidermal KO |
| DKO | Double GR/MR epidermal KO |
| HDAC | Histone deacetylase |
| HSP | Heat shock protein |
| NF-κB | Nuclear factor-kappaB |
| AP-1 | Activator protein 1 |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal regulated kinase |
| SMRT | Silencing mediator for retinoid or thyroid hormone receptors |
References
- Schmuth, M.; Watson, R.E.; Deplewski, D.; Dubrac, S.; Zouboulis, C.C.; Grifiths, C.E. Nuclear hormone receptors in human skin. Horm. Metab. Res. 2007, 39, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J. Ancient friends, revisited: New aspects on the important role of nuclear receptor signalling for skin physiology and for the treatment of skin diseases. Dermatoendocrinology 2011, 3, 121–124. [Google Scholar] [CrossRef]
- Pérez, P. Glucocorticoid receptors, epidermal homeostasis and hair follicle differentiation. Dermatoendocrinology 2011, 3, 1–9. [Google Scholar] [CrossRef]
- Farman, N.; Maubec, E.; Poeggeler, B.; Klatte, J.E.; Jaisser, F.; Paus, R. The mineralocorticoid receptor as a novel player in skin biology: Beyond the renal horizon? Exp. Dermatol. 2010, 19, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Granner, D.K.; Wang, J.C.; Yamamoto, K.R. Regulatory actions of glucocorticoid hormones: From organisms to mechanisms. In Advances in Experimental Medicine and Biology. Glucocorticoid Signaling from Molecules to Mice to Man; Wang, J.C., Harris, C., Eds.; Springer: New York, NY, USA, 2015; pp. 3–31. [Google Scholar]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Martinerie, L.; Munier, M.; Menuet, D.L.; Meduri, G.; Viengchareun, S.; Lombès, M. The mineralocorticoid signaling pathway throughout development: Expression, regulation and pathophysiological implications. Biochimie 2013, 95, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Jaisser, F.; Farman, N. Emerging roles of the mineralocorticoid receptor in pathology: Toward new paradigms in clinical pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Glucocorticoid signaling in the heart: A cardiomyocyte perspective. J. Steroid Biochem. Mol. Biol. 2015, 153, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Latorre, V.; Sanchis, A.; Pérez, P. Epidermal inactivation of the glucocorticoid receptor triggers skin barrier defects and cutaneous inflammation. J. Investig. Dermatol. 2013, 133, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Sevilla, L.M.; Sáez, Z.; Carceller, E.; Pérez, P. Epidermal mineralocorticoid receptor plays beneficial and adverse effects in skin and mediates glucocorticoid responses. J. Investig. Dermatol. 2016, 136, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.U.; Fuchs, E. Skin and its regenerative powers: An alliance between stem cells and their niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. Mammalian skin cell biology: At the interface between laboratory and clinic. Science 2014, 346, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta 2013, 1833, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.I. Making an epidermis. Ann. N. Y. Acad. Sci. 2009, 1170, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Desmet, S.J.; De Bosscher, K. Glucocorticoid receptors: Finding the middle ground. J. Clin. Investig. 2017, 127, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Katsu, Y. 30 years of the mineralocorticoid receptor: Evolution of the mineralocorticoid receptor: Sequence, structure and function. J. Endocrinol. 2017, 234, T1–T16. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Yang, J.; Young, M.J. 30 years of the mineralocorticoid receptor: Coregulators as mediators of mineralocorticoid receptor signalling diversity. J. Endocrinol. 2017, 234, T23–T34. [Google Scholar] [CrossRef] [PubMed]
- Faresse, N. Post-translational modifications of the mineralocorticoid receptor: How to dress the receptor according to the circumstances? J. Steroid Biochem. Mol. Biol. 2014, 143, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Le Tallec, L.; Lombès, M. The mineralocorticoid receptor: A journey exploring its diversity and specificity of action. Mol. Endocrinol. 2005, 19, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Souque, A.; Viengchareun, S.; Poisson, E.; Lombès, M. A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol. Endocrinol. 2001, 15, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Newton, R. Anti-inflammatory glucocorticoids: Changing concepts. Eur. J. Pharmacol. 2013, 724, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; de Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef]
- Presman, D.M.; Ogara, M.F.; Stortz, M.; Alvarez, L.D.; Pooley, J.R.; Schiltz, R.L.; Grøntved, L.; Johnson, T.A.; Mittelstadt, P.R.; Ashwell, J.D.; et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. 2014, 12, e1001813. [Google Scholar] [CrossRef] [PubMed]
- Presman, D.M.; Ganguly, S.; Schiltz, R.L.; Johnson, T.A.; Karpova, T.S.; Hager, G.L. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc. Natl. Acad. Sci. USA 2016, 113, 8236–8241. [Google Scholar] [CrossRef] [PubMed]
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid signaling: An update from a genomic perspective. Ann. Rev. Physiol. 2016, 78, 155–180. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Fujiki, K.; Shirahige, K.; Gomez-Sanchez, CE.; Fujita, T.; Nangaku, M.; Nagase, M. Genome-wide analysis of murine renal distal convoluted tubular cells for the target genes of mineralocorticoid receptor. Biochem. Biophys. Res. Commun. 2014, 445, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Le Billan, F.; Khan, J.A.; Lamribet, K.; Viengchareun, S.; Bouligand, J.; Fagart, J.; Lombès, M. Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 2015, 29, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Van Weert, L.; Buurstede, J.C.; Mahfouz, A.; Braakhuis, P.S.M.; Polman, J.A.E.; Sips, H.C.M.; Roozendaal, B.; Balog, J.; de Kloet, E.R.; Datson, N.A.; et al. NeuroD factors discriminate mineralocorticoid from glucocorticoid receptor DNA binding in the male rat brain. Endocrinology 2017, 158, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- So, A.Y.; Chaivorapol, C.; Bolton, E.C.; Li, H.; Yamamoto, K.R. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007, 3, e94. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Meijsing, S.H.; Pufall, M.A.; So, A.Y.; Bates, D.L.; Chen, L.; Yamamoto, K.R. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 2009, 324, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Schiller, B.J.; Chodankar, R.; Watson, L.C.; Stallcup, M.R.; Yamamoto, K.R. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. 2014, 15, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Radoja, N.; Komine, M.; Jho, S.H.; Blumenberg, M.; Tomic-Canic, M. Novel mechanism of steroid action in skin through glucocorticoid receptor monomers. Mol. Cell. Biol. 2000, 20, 4328–4339. [Google Scholar] [CrossRef] [PubMed]
- Uhlenhaut, N.H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.; Hübner, N.; Evans, R.M. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol. Cell 2013, 49, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Latorre, V.; Carceller, E.; Boix, J.; Vodák, D.; Mills, I.G.; Pérez, P. Glucocorticoid receptor and Klf4 co-regulate anti-inflammatory genes in keratinocytes. Mol. Cell. Endocrinol. 2015, 412, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Ruhs, S.; Langenbruch, L.; Mildenberger, S.; Strätz, N.; Schumann, K.; Gekle, M. Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem. Biol. 2012, 19, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Savory, J.G.; Préfontaine, G.G.; Lamprecht, C.; Liao, M.; Walther, R.F.; Lefebvre, Y.A.; Haché, R.J. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol. Cell. Biol. 2001, 21, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, K.R.; Reul, J.M. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc. Natl. Acad. Sci. USA 2016, 113, 11336–11341. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, K.R.; Reul, J.M.H.M. Mineralocorticoid and glucocorticoid receptor-mediated control of genomic responses to stress in the brain. Stress 2018, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Le Billan, F.; Amazit, L.; Bleakley, K.; Xue, Q.Y.; Pussard, E.; Lhadj, C.; Kolkhof, P.; Viengchareun, S.; Fagart, J.; Lombès, M. Corticosteroid receptors adopt distinct cyclical transcriptional signatures. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bigas, J.; Sevilla, L.M.; Carceller, E.; Boix, J.; Pérez, P. Epidermal glucocorticoid and mineralocorticoid receptors act cooperatively to regulate epidermal development and counteract skin inflammation. Cell Death Dis. 2018, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Ruhs, S.; Nolze, A.; Hübschmann, R.; Grossmann, C. 30 years of the mineralocorticoid receptor: Nongenomic effects via the mineralocorticoid receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef] [PubMed]
- Leis, H.; Page, A.; Ramírez, A.; Bravo, A.; Segrelles, C.; Paramio, J.; Barettino, D.; Jorcano, J.L.; Pérez, P. Glucocorticoid Receptor Counteracts Tumorigenic Activity of Akt in Skin through Interference with the Phosphatidylinositol 3-Kinase Signaling Pathway. Mol. Endocrinol. 2004, 18, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Blendy, A.P.; Monaghan, K.; Schmid, W.; Aguzzi, A.; Fantuzzi, G.; Hummler, E.; Unsicker, K.; Schütz, G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995, 9, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Walker, E.A.; Hardy, R.S.; Mayes, A.E.; Stewart, P.M. Localization, age- and site-dependent expression, and regulation of 11β-hydroxysteroid dehydrogenase type 1 in skin. J. Investig. Dermatol. 2011, 131, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Katayama, I. Local cortisol/corticosterone activation in skin physiology and pathology. J. Dermatol. Sci. 2016, 84, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, N.; Prime, S.S. Keratinocytes synthesize and activate cortisol. J. Cell. Biochem. 2011, 112, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Carceller, E.; Sevilla, L.M.; Marcos-Garcés, V.; Pérez, P. The mineralocorticoid receptor plays a transient role in mouse skin development. Exp. Dermatol. 2016, 25, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Bigas, J.; Sevilla, L.M.; Iacobone, M.; Citton, M.; Torresan, F.; Caroccia, B.; Rossi, G.P.; Pérez, P. Primary aldosteronism patients show skin alterations and abnormal activation of glucocorticoid receptor in keratinocytes. Sci. Rep. 2017, 7, 15806. [Google Scholar] [CrossRef] [PubMed]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef] [PubMed]
- Nikolakis, G.; Stratakis, C.A.; Kanaki, T.; Slominski, A.; Zouboulis, C.C. Skin steroidogenesis in health and disease. Rev. Endocr. Metab. Disord. 2016, 17, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Manna, P.R.; Tuckey, R.C. Cutaneous glucocorticosteroidogenesis: Securing local homeostasis and the skin integrity. Exp. Dermatol. 2014, 23, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.K.; Kaplan, N.; Tsoi, L.C.; Xing, X.; Liang, Y.; Swindell, W.R.; Hoover, P.; Aravind, M.; Baida, G.; Clark, M.; et al. Endogenous Glucocorticoid Deficiency in Psoriasis Promotes Inflammation and Abnormal Differentiation. J. Investig. Dermatol. 2017, 137, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Hannen, R.; Udeh-Momoh, C.; Upton, J.; Wright, M.; Michael, A.; Gulati, A.; Rajpopat, S.; Clayton, N.; Halsall, D.; Burrin, J.; et al. Dysfunctional skin-derived glucocorticoid synthesis is a pathogenic mechanism of psoriasis. J. Investig. Dermatol. 2017, 137, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Bleich, M.; Schmid, W.; Cole, T.J.; Peters, J.; Watanabe, H.; Kriz, W.; Warth, R.; Greger, R.; Schütz, G. Mineralocorticoid receptor knockout mice: Pathophysiology of Na+ metabolism. Proc. Natl. Acad. Sci. USA 1998, 95, 9424–9429. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Ann. Dermatol. 2010, 22, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Duan, E. Epidermal development in mammals: Key regulators, signals from beneath, and stem cells. Int. J. Mol. Sci. 2013, 14, 10869–10895. [Google Scholar] [CrossRef] [PubMed]
- Hardman, M.J.; Sisi, P.; Banbury, D.N.; Byrne, C. Patterned acquisition of skin barrier function during development. Development 1998, 125, 1541–1552. [Google Scholar] [PubMed]
- Sevilla, L.M.; Bayo, P.; Latorre, V.; Sanchis, A.; Pérez, P. Glucocorticoid receptor regulates overlapping and differential gene subsets in developing and adult skin. Mol. Endocrinol. 2010, 24, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Makhanova, N.; Caron, K.; Lopez, M.L.; Gomez, R.A.; Smithies, O.; Kim, H.S. Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 2005, 146, 2650–2656. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Feingold, K.R.; Menon, G.K.; Williams, M.L. Glucocorticoids accelerate fetal maturation of the epidermal permeability barrier in the rat. J. Clin. Investig. 1993, 91, 2703–2708. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.; Feingold, K.R.; Kömüves, L.G.; Elias, P.M.; Muglia, L.J.; Majzoub, J.A.; Williams, M.L. Glucocorticoid deficiency delays stratum corneum maturation in the fetal mouse. J. Investig. Dermatol. 1998, 111, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Xi, Z.F.; Seo, E.Y.; McGaughey, D.; Segre, J.A. Klf4 and corticosteroids activate an overlapping set of transcriptional targets to accelerate in utero epidermal barrier acquisition. Proc. Natl. Acad. Sci. USA 2006, 103, 18668–18673. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, A.; Bravo, A.; Jorcano, J.L.; Vidal, M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a LACZ gene in the adult and during development. Differentiation 1994, 58, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; Page, A.; Bravo, A.; del Río, M.; Giménez-Conti, I.; Budunova, I.; Slaga, T.J.; Jorcano, J.L. Altered skin development and impaired proliferative and inflammatory responses in transgenic mice overexpressing the glucocorticoid receptor. FASEB J. 2001, 15, 2030–2032. [Google Scholar] [CrossRef] [PubMed]
- Sainte Marie, Y.; Toulon, A.; Paus, R.; Maubec, E.; Cherfa, A.; Grossin, M.; Descamps, V.; Clemessy, M.; Gasc, J.M.; Peuchmaur, M.; et al. Targeted skin overexpression of the mineralocorticoid receptor in mice causes epidermal atrophy, premature skin barrier formation, eye abnormalities, and alopecia. Am. J. Pathol. 2007, 171, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Cascallana, J.L.; Bravo, A.; Donet, E.; Leis, H.; Lara, M.F.; Paramio, J.M.; Jorcano, J.L.; Pérez, P. Ectoderm-targeted overexpression of the glucocorticoid receptor induces hypohidrotic ectodermal dysplasia. Endocrinology 2005, 146, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Budunova, I.V.; Carbajal, S.; Kang, H.; Viaje, A.; Slaga, T.J. Altered glucocorticoid receptor expression and function during mouse skin carcinogenesis. Mol. Carcinog. 1997, 18, 177–185. [Google Scholar] [CrossRef]
- Stojadinovic, O.; Lee, B.; Vouthounis, C.; Vukelic, S.; Pastar, I.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: Inhibition of apoptosis, interferon-γ pathway, and wound healing along with promotion of terminal differentiation. J. Biol. Chem. 2007, 282, 4021–4034. [Google Scholar] [CrossRef] [PubMed]
- Bayo, P.; Sanchis, A.; Bravo, A.; Cascallana, J.L.; Buder, K.; Tuckermann, J.; Schütz, G.; Pérez, P. Glucocorticoid receptor is required for skin barrier competence. Endocrinology 2008, 149, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Bleich, M.; Warth, R.; Schmidt-Hieber, M.; Schulz-Baldes, A.; Hasselblatt, P.; Fisch, D.; Berger, S.; Kunzelmann, K.; Kriz, W.; Schütz, G.; et al. Rescue of the mineralocorticoid receptor knock-out mouse. Pflug. Arch. 1999, 438, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Bleich, M.; Schmid, W.; Greger, R.; Schütz, G. Mineralocorticoid receptor knockout mice: Lessons on Na+ metabolism. Kidney Int. 2000, 57, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights from Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.K.; Li, Z.J.; Choi, D.K.; Sohn, K.C.; Lim, E.H.; Lee, Y.H.; Kim, S.; Im, M.; Lee, Y.; Seo, Y.J.; et al. Glucocorticoid receptor enhances involucrin expression of keratinocyte in a ligand-independent manner. Mol. Cell. Biochem. 2014, 390, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Latorre, V.; Sevilla, L.M.; Sanchis, A.; Pérez, P. Selective ablation of glucocorticoid receptor in mouse keratinocytes increases susceptibility to skin tumorigenesis. J. Investig. Dermatol. 2013, 133, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Lamprokostopoulou, A.; Sertedaki, A.; Charmandari, E. Recent advances in the molecular mechanisms causing primary generalized glucocorticoid resistance. Hormones 2016, 15, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Schoepe, S.; Schäcke, H.; May, E.; Asadullah, K. Glucocorticoid therapy-induced skin atrophy. Exp. Dermatol. 2006, 15, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Lause, M.; Kamboj, A.; Fernandez-Faith, E. Dermatologic manifestations of endocrine disorders. Transl. Pediatr. 2017, 6, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Schottelius, A.; Döcke, W.D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc. Natl. Acad. Sci. USA 2004, 101, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.; Yemelyanov, A.; Swart, A.C.; Swart, P.; Haegeman, G.; Budunova, I. Discovery of Compound A-a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 2015, 6, 30730–30744. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; de Bosscher, K.; Libert, C. The interactome of the glucocorticoid receptor and its influence on the actions of glucocorticoids in combatting inflammatory and infectious diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Perera, D.N.; Fan, H.; Russ, B.E.; Harris, J.; Morand, E.F. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J. Autoimmun. 2015, 61, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Carceller, E.; Ballegeer, M.; Deckers, J.; Riccardi, C.; Bruscoli, S.; Hochepied, T.; Libert, C.; Pérez, P. Overexpression of glucocorticoid-induced Leucine Zipper (GILZ) increases susceptibility to imiquimod-induced psoriasis and involves cutaneous activation of TGF-β1. Sci. Rep. 2016, 6, 38825. [Google Scholar] [CrossRef] [PubMed]
- Farman, N.; Nguyen, V.T. A novel actor in skin biology: The mineralocorticoid receptor. Exp. Dermatol. 2016, 25, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Laouénan, C.; Deschamps, L.; Nguyen, V.T.; Scheer-Senyarich, I.; Wackenheim-Jacobs, A.C.; Steff, M.; Duhamel, S.; Tubiana, S.; Brahimi, N.; et al. Topical mineralocorticoid receptor blockade limits glucocorticoid-induced epidermal atrophy in human skin. J. Investig. Dermatol. 2015, 135, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Nguyen, V.T.; Farman, N.; Aractingi, S.; Pérez, P. Mineralocorticoid receptor blockade improves glucocorticoid-induced skin atrophy but partially ameliorates anti-inflammatory actions in an irritative model in human skin explants. Exp. Dermatol. 2017, 27, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ghadially, R.; Brown, B.E.; Sequeira-Martin, S.M.; Feingold, K.R.; Elias, P.M. The aged epidermal permeability barrier. Structural, functional, and lipid biochemical abnormalities in humans and a senescent murine model. J. Clin. Investig. 1995, 95, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef] [PubMed]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in wound healing: A comprehensive review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Vukelic, S.; Stojadinovic, O.; Pastar, I.; Rabach, M.; Krzyzanowska, A.; Lebrun, E.; Davis, S.C.; Resnik, S.; Brem, H.; Tomic-Canic, M. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J. Biol. Chem. 2011, 286, 10265–10275. [Google Scholar] [CrossRef] [PubMed]
- Youm, J.K.; Park, K.; Uchida, Y.; Chan, A.; Mauro, T.M.; Holleran, W.M.; Elias, P.M. Local blockade of glucocorticoid activation reverses stress- and glucocorticoid-induced delays in cutaneous wound healing. Wound Repair Regen. 2013, 21, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Tahrani, A.A.; Morgan, S.A.; Otranto, M.; Desmoulière, A.; Abrahams, L.; Hassan-Smith, Z.; Walker, E.A.; Rabbitt, E.H.; Cooper, M.S.; et al. 11β-Hydroxysteroid dehydrogenase blockade prevents age-induced skin structure and function defects. J. Clin. Investig. 2013, 123, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, A.; Alba, L.; Latorre, V.; Sevilla, L.M.; Pérez, P. Keratinocyte-targeted overexpression of the glucocorticoid receptor delays cutaneous wound healing. PLoS ONE 2012, 7, e29701. [Google Scholar] [CrossRef] [PubMed]
- Donet, E.; Bosch, P.; Sanchis, A.; Bayo, P.; Ramírez, A.; Cascallana, J.L.; Bravo, A.; Pérez, P. Transrepression function of the glucocorticoid receptor regulates eyelid development and keratinocyte proliferation but is not sufficient to prevent skin chronic inflammation. Mol. Endocrinol. 2008, 22, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, O.; Brem, H.; Vouthounis, C.; Lee, B.; Fallon, J.; Stallcup, M.; Merchant, A.; Galiano, R.D.; Tomic-Canic, M. Molecular pathogenesis of chronic wounds: The role of β-catenin and c-Myc in the inhibition of epithelialization and wound healing. Am. J. Pathol. 2005, 167, 59–69. [Google Scholar] [CrossRef]
- Sawaya, A.P.; Pastar, I.; Stojadinovic, O.; Lazovic, S.; Davis, S.C.; Gil, J.; Kirsner, R.S.; Tomic-Canic, M. Topical mevastatin promotes wound healing by inhibiting the transcription factor c-Myc via the glucocorticoid receptor and the long non-coding RNA Gas5. J. Biol. Chem. 2018, 293, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Jozic, I.; Vukelic, S.; Stojadinovic, O.; Liang, L.; Ramirez, H.A.; Pastar, I.; Tomic Canic, M. Stress signals, mediated by membranous glucocorticoid receptor, activate PLC/PKC/GSK-3β/β-catenin pathway to inhibit wound closure. J. Investig. Dermatol. 2017, 137, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A. Glucocorticoids inhibit wound healing: Novel mechanism of action. J. Investig. Dermatol. 2017, 137, 1012–1014. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Farman, N.; Maubec, E.; Nassar, D.; Desposito, D.; Waeckel, L.; Aractingi, S.; Jaisser, F. Re-epithelialization of pathological cutaneous wounds is improved by local mineralocorticoid receptor antagonism. J. Investig. Dermatol. 2016, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Akase, T.; Sanada, H.; Minematsu, T.; Ibuki, A.; Huang, L.; Asada, M.; Yoshimura, K.; Nagase, M.; Shimada, T.; et al. Aging-like skin changes in metabolic syndrome model mice are mediated by mineralocorticoid receptor signaling. Aging Cell 2013, 12, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bollag, W.B.; Isales, C.M. GRowing an epidermal tumor. J. Investig. Dermatol. 2013, 133, 2659–2662. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Paulen, L.; Chambon, P. GR SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc. Natl. Acad. Sci. USA 2016, 113, E626–E634. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Ganti, K.P.; Chambon, P. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc. Natl. Acad. Sci. USA 2016, 113, E635–E643. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Canino, R.; Fernandes, M.X. Alvarez de la Rosa, D. Phosphorylation of mineralocorticoid receptor ligand binding domain impairs receptor activation and has a dominant negative effect over non-phosphorylated receptors. J. Biol. Chem. 2016, 291, 19068–19078. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fariñas, M.; Li, K.; Fuentes-Duculan, J.; Hayden, K.; Brodmerkel, C.; Krueger, J.G. Expanding the psoriasis disease profile: Interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J. Investig. Dermatol. 2012, 132, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevilla, L.M.; Pérez, P. Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology. Int. J. Mol. Sci. 2018, 19, 1906. https://doi.org/10.3390/ijms19071906
Sevilla LM, Pérez P. Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology. International Journal of Molecular Sciences. 2018; 19(7):1906. https://doi.org/10.3390/ijms19071906
Chicago/Turabian StyleSevilla, Lisa M., and Paloma Pérez. 2018. "Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology" International Journal of Molecular Sciences 19, no. 7: 1906. https://doi.org/10.3390/ijms19071906
APA StyleSevilla, L. M., & Pérez, P. (2018). Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology. International Journal of Molecular Sciences, 19(7), 1906. https://doi.org/10.3390/ijms19071906