Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Effect of Pb on Cytokines
2.1. Interleukins 2 and 4 (IL-2 and IL-4)
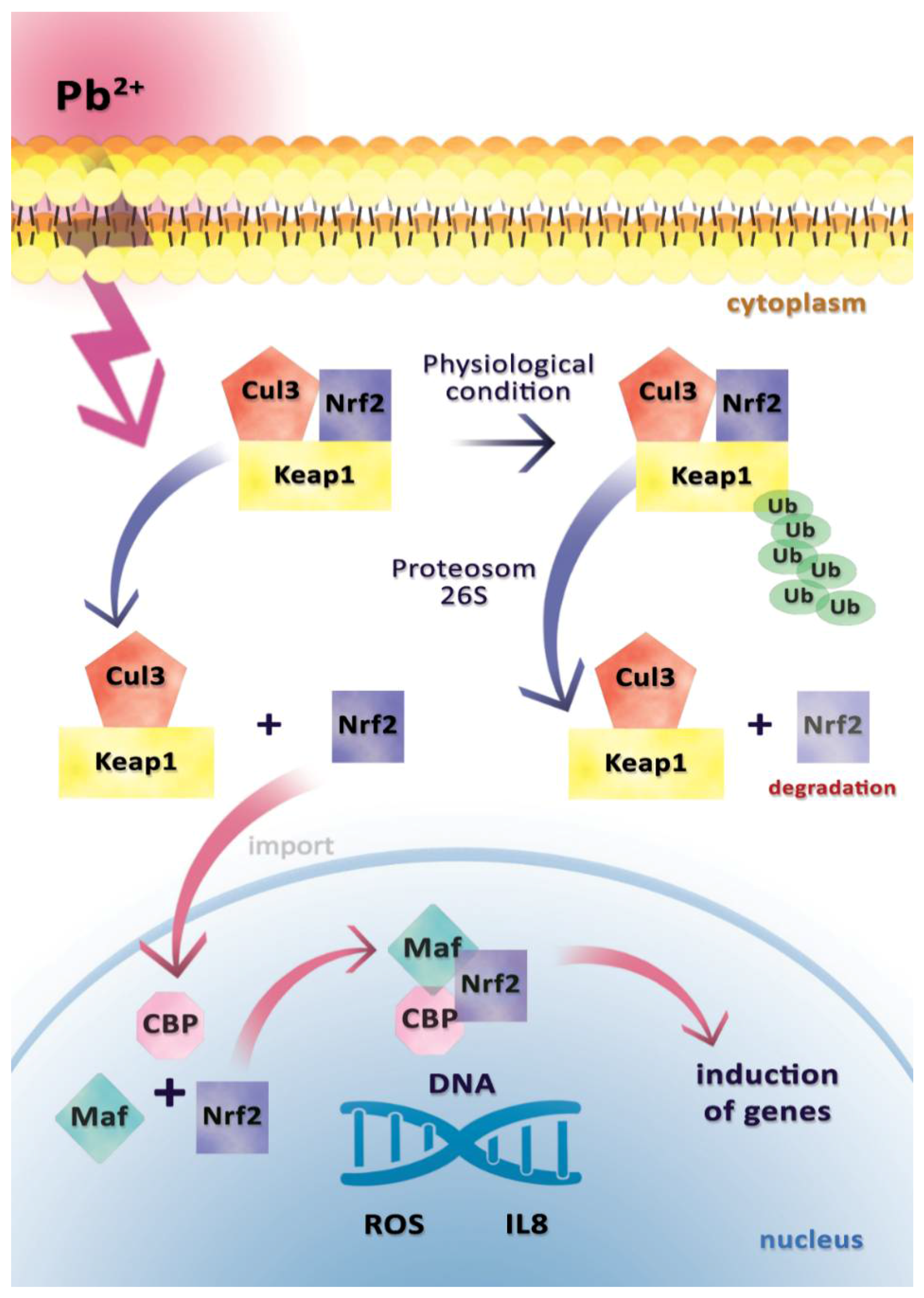
2.2. Interleukin 8 (IL-8)
2.3. Interleukins 1b and 6 (IL-1b and IL-6)
2.4. Interferon Gamma (IFNγ)
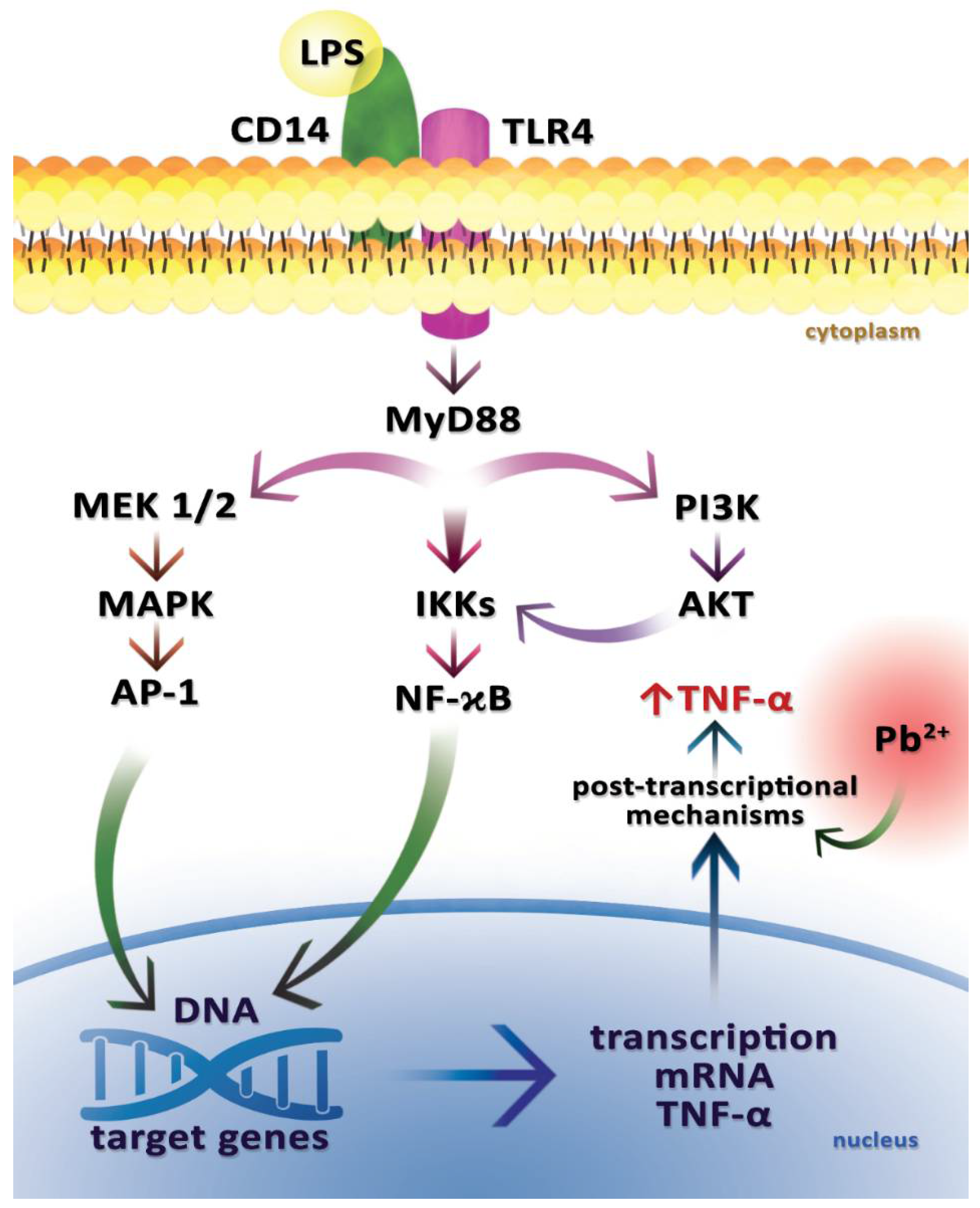
2.5. Tumor Necrosis Factor Alpha (TNF-α)
3. Influence of Pb on Enzymes Involved in Inflammation
3.1. Cyclooxygenase 2 (COX-2)
3.2. Lipoxygenases
4. Effect of Pb on Selected Acute Phase Proteins: C-Reactive Protein (CRP), Haptoglobin, Ceruloplasmin
4.1. CRP
4.2. Haptoglobin
4.3. Ceruloplasmin
5. Influence of Pb on Immune System Cells
5.1. The Effect of Pb on Lymphocytes T, B, and NK
5.2. Effect of Pb on Macrophages
5.3. Effect of Pb on Dendritic Cells
6. The Effect of Pb on Immunoglobulins (IgA, IgG, IgM)
7. The Effect of Pb on Histamine, IgE, and Endothelin
7.1. Histamine and Immunoglobin E
7.2. Endothelin
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- ATSDR (Agency for Toxic Substances and Disease Registry’s). CERCLA Priority List of Most Hazardous Substances. 2007. Available online: http://www.atsdr.cdc.gov/cercla (accessed on 1 September 2017).
- EP (Environmental Protection). The Restriction of the Use of Certain Hazardous Substances in Electrical and Electronic Equipment Regulations; No. 2748. 2005. Available online: http://www.legislation.gov.uk/uksi/2005/2748/pdfs/uksi_20052748_en.pdf (accessed on 25 September 2017).
- EP (Environmental Protection). The Restriction of the Use of Certain Hazardous Substances in Electrical and Electronic Equipment (Amendment) Regulations; No. 581. 2009. Available online: http://www.legislation.gov.uk/uksi/2009/581/pdfs/uksi_20090581_en.pdf (accessed on 25 October 2017).
- EU European Commission Institute for Health and Consumer Protection. Toxicology and Chemical Substances (& ECB): Opinion of the TC NES on the Environment Part of Industry Voluntary Risk Assessments on Lead and Lead Compounds. 2008. Available online: http://echa.europa.eu/doc/trd_substances/VRAR/Lead/tcnes_opinion/tcnes_opinion_env.pdf (accessed on 25 September 2017).
- World Health Organization (WHO). Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks; World Health Organization: Geneva, Switzerland, 2009; Available online: http://www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf (accessed on 25 September 2017).
- Skoczyńska, A.; Poręba, R.; Sieradzki, A.; Andrzejak, R.; Sieradzka, U. Wpływ ołowiu i kadmu na funkcje układu immunologicznego. Med. Pr. 2002, 53, 259–264. [Google Scholar] [PubMed]
- Schuster, H.P. Intensywna Terapia w Posocznicy, 1st ed.; Sanmedica: Warszawa, Poland, 1997; ISBN 83-86516-29-1. [Google Scholar]
- Ramella, M.; Boccafoschi, F.; Bellofatto, K.; Md, A.F.; Fusaro, L.; Boldorini, R.; Casella, F.; Porta, C.; Settembrini, P.; Cannas, M. Endothelial MMP-9 drives the inflammatory response in abdominal aortic aneurysm (AAA). Am. J. Transl. Res. 2017, 9, 5485–5495. [Google Scholar] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Piepenbrink, M.S. Lead and Immune Function. Crit. Rev. Toxicol. 2006, 36, 359–385. [Google Scholar] [CrossRef] [PubMed]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int. J. Mol. Sci. 2016, 17, 2140. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Oxenius, A. Interleukin 2: From immunostimulation to immunoregulation and back again. EMBO Rep. 2007, 8, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.; Reiser, H. IL-4: Role in disease and regulation of production. Clin. Exp. Immunol. 1998, 113, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Iavicoli, I.; Carelli, G.; Stanek, E.J., 3rd; Castellino, N.; Calabrese, E.J. Below background levels of blood lead impact cytokine levels in male and female mice. Toxicol. Appl. Pharmacol. 2006, 210, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Wei, P.L.; Tsai, Y.T.; Wong, J.H.; Chang, C.M.; Wang, J.Y.; Hou, M.F.; Lee, Y.C.; Chuang, H.Y.; Chang, W.C. Pb2+ induced IL-8 gene expression by extracellular signal-regulated kinases and the transcription factor, activator protein 1, in human gastric carcinoma cells. Environ. Toxicol. 2015, 30, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Yu, M.C.; Chiang, C.C.; Bien, M.Y.; Chien, M.H.; Chen, B.C. Thrombin-induced NF-kappaB activation and IL-8/CXCL8 release is mediated by c-Src-dependent Shc, Raf-1, and ERK pathways in lung epithelial cells. Cell. Signal. 2013, 25, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar] [PubMed]
- Chou, Y.H.; Woon, P.Y.; Huang, W.C.; Shiurba, R.; Tsai, Y.T.; Wang, Y.S.; Hsieh, T.J.; Chang, W.C.; Chuang, H.Y.; Chang, W.C. Divalent lead cations induce cyclooxygenase-2 gene expression by epidermal growth factor receptor/nuclear factor-kappa B signaling in A431carcinoma cells. Toxicol. Lett. 2011, 203, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.C.; Chang, C.C.; Wang, Y.S.; Weng, W.T.; Yoshioka, T.; Juo, S.H. Involvement of the epidermal growth factor receptor in Pb2+-induced activation of cPLA2/COX-2 genes and PGE2 production in vascular smooth muscle cells. Toxicology 2011, 279, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Gillis, B.S.; Arbieva, Z.; Gavin, I.M. Analysis of lead toxicity in human cells. BMC Genom. 2012, 13, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Zeller, I.; Knoflach, M.; Seubert, A.; Kreutmayer, S.B.; Stelzmuller, M.E.; Wallnoefer, E.; Blunder, S.; Frotschnig, S.; Messner, B.; Willeit, J.; et al. Lead contributes to arterial intimal hyperplasia through nuclear factor erythroid 2-related factor-mediated endothelial interleukin 8 synthesis and subsequent invasion of smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A. Regulation of interleukin-8 gene expression. J. Interferon Cytokine Res. 1999, 19, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Fu, Y.; Yang, H. Leptin and IL-8: Two novel cytokines screened out in childhood lead exposure. Toxicol. Lett. 2014, 227, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Roper, W.L.; Houk, V.; Falk, H.; Binder, S. Preventing Lead Poisoning in Young Children: A Statement by the Centers for Disease Control, October 1991; Centers for Disease Control: Atlanta, GA, USA, 1991; Available online: http://www.nmic.org/nyccelp/medical-studies/CDC-Preventing-lead-poisoning-10-91.pdf (accessed on 1 September 2017).
- CDC. Low Level Lead Exposure Harms Children: A Renewed Call for Primary Prevention. Report of the Advisory Committee on Childhood Lead Poisoning Prevention of the Centers for Disease Control and Prevention; 2012. Available online: https://www.cdc.gov/nceh/lead/acclpp/final_document_030712.pdf (accessed on 25 July 2017).
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Dyatlov, V.A.; Lawrence, D.A. Neonatal Lead Exposure Potentiates Sickness Behavior Induced by Listeria monocytogenes Infection of Mice. Brain Behav. Immun. 2002, 16, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.E.; Filipov, N.M.; Parsons, P.J.; Lawrence, D.A. The Efficiency of Maternal Transfer of Lead and Its Influence on Plasma IgE and Splenic Cellularity of Mice. Toxicol. Sci. 2000, 57, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Needleman, H. Lead poisoning. Annu. Rev. Med. 2004, 55, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Heo, Y.; Lawrence, D.A. Central nervous system cytokine gene expression: Modulation by lead. J. Biochem. Mol. Toxicol. 2011, 25, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Klasen, H.J.; Imfeld, K.L.; Kirov, I.I.; Tai, L.; Gage, F.H.; Young, M.J.; Berman, M.A. Expression of cytokines by multipotent neural progenitor cells. Cytokine 2003, 22, 101–106. [Google Scholar] [CrossRef]
- Kishikawa, H.; Lawrence, D.A. Differential production of interleukin-6 in the brain and spleen of mice treated with lipopolysaccharide in the presence and absence of lead. J. Toxicol. Environ. Health Part A 1998, 53, 357–373. [Google Scholar] [PubMed]
- Vagaska, B.; New, S.E.P.; Alvarez-Gonzalez, C.; D’Acquisto, F.; Gomez, S.G.; Bulstrode, N.W.; Madrigal, A.; Ferretti, P. MHC-class-II are expressed in a subpopulation of human neural stem cells in vitro in an IFNγ–independent fashion and during development. Sci. Rep. 2016, 6, 24251. [Google Scholar] [CrossRef] [PubMed]
- Yücesoy, B.; Turhan, A.; Ure, M.; Imir, T.; Karakaya, A. Effects of occupational lead and cadmium exposure on some immunoregulatory cytokine levels in man. Toxicology 1997, 123, 143–147. [Google Scholar] [CrossRef]
- Heo, Y.; Parsons, P.J.; Lawrence, D.A. Lead differentially modifies cytokine production in vitro and in vivo. Toxicol. Appl. Pharmacol. 1996, 138, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, H.; Song, R.; Lawrence, D.A. Interleukin-12 promotes enhanced resistance to Listeria monocytogenes infection of lead-exposed mice. Toxicol. Appl. Pharmacol. 1997, 147, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Golemboski, K.A.; Ha, R.S.; Bunn, T.; Sanders, F.S.; Dietert, R.R. Developmental exposure to lead causes persistent immunotoxicity in Fischer 344 rats. Toxicol. Sci. 1998, 42, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.; Lee, W.T.; Lawrence, D.A. Differential effects of lead and cAMP on development and activities of Th1- and Th2-lymphocytes. Toxicol. Sci. 1998, 43, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.; Mondal, T.K.; Gao, D.; Kasten-Jolly, J.; Kishikawa, H.; Lawrence, D.A. Posttranscriptional inhibition of interferon-gamma production by lead. Toxicol. Sci. 2007, 96, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, T.; Filar, J.; Madej, E.; Szuster-Ciesielska, A.; Kandefer-Szerszeń, M. Modification of bovine interferon and tumor necrosis factor production by lead in vivo and in vitro. Arch. Immunol. Ther. Exp. 1998, 46, 323–328. [Google Scholar]
- Radbin, R.; Vahedi, F.; Chamani, J. The influence of drinking-water pollution with heavy metal on the expression of IL-4 and IFNγ in mice by real-time polymerase chain reaction. Cytotechnology 2014, 66, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.L.; Mudzinski, S.P.; Lawrence, D.A. The heavy metal lead modulates the expression of both TNF-alpha and TNF-alpha receptors in lipopolysaccharide-activated human peripheral blood mononuclear cells. J. Leukoc. Biol. 1996, 59, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Long, G.J.; Rosen, J.F.; Schanne, F.A. Lead activation of protein kinase C from rat brain. J. Biol. Chem. 1994, 269, 834–837. [Google Scholar] [PubMed]
- Erickson, S.L.; de Sauvage, F.J.; Kikly, K.; Carver-Moore, K.; Pitts-Meek, S.; Gillett, N.; Sheehan, K.C.F.; Schreiber, R.D.; Goeddel, D.V.; Moore, M.W. Decreased sensitivity to tumor-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.; Evans, T.J.; Buurman, W.A.; Bemelmans, M.H.; Moyes, D.; Cohen, J. Differences in the shedding of soluble TNF receptors between endotoxin-sensitive and endotoxin-resistant mice in response to lipopolysaccharide or live bacterial challenge. J. Immunol. 1995, 155, 2005–2012. [Google Scholar] [PubMed]
- Goldfeld, A.E.; Doyle, C.; Maniatis, T. Human tumor necrosis factor alpha gene regulation by virus and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1990, 87, 9769–9773. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.J.; Yang, B.C.; Liu, M.Y. Lead increases lipopolysaccharide-induced liver-injury through tumor necrosis factor-α overexpression by monocytes/macrophages: Role of protein kinase C and P42/44 mitogen-activated protein kinase. Environ. Health Perspect. 2006, 114, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Dentener, M.A.; Greve, J.W.; Maessen, J.G.; Buurman, W.A. Role of tumour necrosis factor in the enhanced sensitivity of mice to endotoxin after exposure to lead. Immunopharmacol. Immunotoxicol. 1989, 11, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Hofsli, E.; Bakke, O.; Nonstad, U.; Espevik, T. A flow cytometnc and immunofluorescence microscopic study of tumor necrosis factor production and localization in human monocytes. Cell. Immunol. 1989, 122, 405–415. [Google Scholar] [CrossRef]
- Hewett, J.A.; Jean, P.A.; Kunkel, S.L.; Roth, R.A. Relationship between tumor necrosis factor-alpha and neutrophils in endotoxin-induced liver injury. Am. J. Physiol. 1993, 265, G1011–G1015. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Gantner, F.; Künstle, G.; Bohlinger, I.; Tiegs, G.; Bluethmann, H.; Wendel, A. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol. Med. 1995, 2, 109–124. [Google Scholar]
- Schuchmann, M.; Varfolomeev, E.E.; Hermann, F.; Rueckert, F.; Strand, D.; Koehler, H.; Strand, S.; Lohse, A.W.; Wallach, D.; Galle, P.R. Dominant negative MORT1/FADD rescues mice from CD95 and TNF-induced liver failure. Hepatology 2003, 37, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Künstle, G.; Leist, M.; Uhlig, S.; Revesz, L.; Feifel, R.; MacKenzie, A.; Wendel, A. ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol. Lett. 1997, 55, 5–10. [Google Scholar] [CrossRef]
- Comalada, M.; Xaus, J.; Valledor, A.F.; Lopez-Lopez, C.; Pennington, D.J.; Celada, A. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. Am. J. Physiol. Cell-Physiol. 2003, 285, C1235–C1245. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Cerami, A. The biology of cachectin/TNF—A primary mediator of the host response. Annu. Rev. 1989, 7, 625–655. [Google Scholar] [CrossRef] [PubMed]
- Appleby, S.B.; Ristimaki, A.; Neilson, K.; Narko, K.; Hla, T. Structure of the human cyclo-oxygenase-2 gene. Biochem. J. 1994, 302, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Yokoyama, C.; Hara, S.; Tone, Y.; Tanabe, T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J. Biol. Chem. 1995, 270, 24965–24971. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ji, Y.S.; Schmedtje, J.F., Jr. Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium, Implications for the mechanisms of aortic aneurysm and heart failure. J. Biol. Chem. 2000, 275, 24583–24589. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.R.; Aguado, A.; Fiorim, J.; Silveira, E.A.; Azevedo, B.F.; Toscano, C.M.; Zhenyukh, O.; Briones, A.M.; Alonso, M.J.; Vassallo, D.V.; et al. MAPK pathway activation by chronic lead-exposure increases vascular reactivity through oxidative stress/cyclooxygenase-2-dependent pathways. Toxicol. Appl. Pharmacol. 2015, 283, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Ohnaka, K.; Numaguchi, K.; Yamakawa, T.; Inagami, T. Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension 2000, 35, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xi, S.; Xu, Y.; Wang, F.; Zheng, Y.; Li, B.; Li, X.; Zheng, Q.; Sun, G. Sodium arsenite induces cyclooxygenase-2 expression in human uroepithelial cells through MAPK pathway activation and reactive oxygen species induction. Toxicol. In Vitro 2013, 27, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.T.; Chang, C.M.; Wang, J.Y.; Hou, M.F.; Wang, J.M.; Shiurba, R.; Chang, W.C.; Chang, W.C. Function of DNA Methyltransferase 3a in Lead (Pb2+)-Induced Cyclooxygenase-2 Gene. Environ. Toxicol. 2015, 30, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Walther, M.; Kuban, R.J. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002, 68–69, 263–290. [Google Scholar] [CrossRef]
- Chaitidis, P.; O’Donnell, V.; Kuban, R.J.; Bermudez-Fajardo, A.; Ungethuem, U.; Kuhn, H. Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and -13. Cytokine 2005, 30, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.J.; Kuhn, H.; Mulkins, M.; Highland, E.; Sigal, E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc. Natl. Acad. Sci. USA 1992, 89, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, J.; Hersberger, M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Shureiqi, I.; Lippman, S.M. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001, 61, 6307–6312. [Google Scholar] [PubMed]
- Poeckel, D.; Zemski Berry, K.A.; Murphy, R.C.; Funk, C.D. Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in ApoE knock-out mice. J. Biol. Chem. 2009, 284, 21077–21089. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Colangelo, V.; Lukiw, W.J. Prostaglandins and other lipid mediators in Alzheimer’s disease. Prostaglandins Other Lipid Mediat. 2002, 68–69, 197–210. [Google Scholar] [CrossRef]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M.; Berglund, M.; Akesson, A.; Lidén, C. Metals and women’s health. Environ. Res. 2002, 88, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic metals and oxidative stress part I: Mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.Z.; Schisterman, E.F.; Goldman, L.R.; Mumford, S.L.; Perkins, N.J.; Bloom, M.S.; Rudra, C.B.; Browne, R.W.; Wactawski-Wende, J. Relation of Blood Cadmium, Lead, and Mercury Levels to Biomarkers of Lipid Peroxidation in Premenopausal Women. Am. J. Epidemiol. 2012, 175, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Songdej, N.; Winters, P.C.; McCabe, M.J., Jr.; van Wijngaarden, E. A population-based assessment of blood lead levels in relation to inflammation. Environ. Res. 2010, 110, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Kasperczyk, A.; Kasperczyk, S.; Horak, S.; Ostałowska, A.; Grucka-Mamczar, E.; Romuk, E.; Olejek, A.; Birkner, E. Assessment of semen function and lipid peroxidation among lead exposed men. Toxicol. Appl. Pharmacol. 2008, 228, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Boncler, M.; Luzak, B.; Watała, C. Znaczenie białka C-reaktywnego w patofizjologii miażdżycy. Role of C-reactive protein in atherogenesis. Postepy Hig. Med. Dosw. 2006, 60, 538–546. [Google Scholar]
- Khan, D.A.; Qayyum, S.; Saleem, S.; Khan, F.A. Lead-induced oxidative stress adversely affects health of the occupational workers. Toxicol. Ind. Health 2008, 24, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, A. Choroby Wewnętrzne. Tom II; Wydawnictwo Medycyna Praktyczna: Kraków, Poland, 2005; p. 111. ISBN 83-7430-031-0. [Google Scholar]
- Kasperczyk, A.; Prokopowicz, A.; Dobrakowski, M.; Pawlas, N.; Kasperczyk, S. The Effect of Occupational Lead Exposure on Blood Levels of Zinc, Iron, Copper, Selenium and Related Proteins. Biol. Trace Elem. Res. 2012, 150, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Luster, M.I.; Faith, R.E.; Kimmel, C.A. Depression of humoral immunity in rats following chronic developmental lead exposure. J. Environ. Pathol. Toxicol. 1978, 1, 397–402. [Google Scholar] [PubMed]
- Hemphil, F.E.; Kaeberle, M.L.; Buck, W.B. Lead suppression of mouse resistance to Salmonella typhimurium. Science 1971, 172, 1031–1032. [Google Scholar] [CrossRef]
- Gainer, J.H. Lead Aggravates Viral Disease and Represses the Antiviral Activity of Interferon Inducers. Environ. Health Perspect. 1974, 7, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Desai, D.; Weiss, A. The role of tyrosine kinases and protein tyrosine phosphatases in the antigen receptor signal transduction. Annu. Rev. Immunol. 1994, 12, 555–592. [Google Scholar] [CrossRef] [PubMed]
- Cambier, J.C.; Pleiman, C.M.; Clark, M.R. Signal transduction by the B cell antigen receptor and its co-receptors. Annu. Rev. Immunol. 1994, 12, 457–486. [Google Scholar] [CrossRef] [PubMed]
- Robey, E.; Allison, J.P. T cell activation: Integration of signals from the antigen receptor and costimulatory molecules. Immunol. Today 1995, 16, 306–310. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signaling. Ann. N. Y. Acad. Sci. 1995, 766, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.R.; Clipstone, N.A. Signal transmission between the plasma membrane and nucleus of T-lymphocytes. Annu. Rev. Biochem. 1994, 63, 1045–1083. [Google Scholar] [CrossRef] [PubMed]
- Razani-Boroujerdi, S.; Edwards, B.; Sopori, M.L. Lead stimulate lymphocyte proliferation through enhanced T cell-B cell interaction. J. Pharmacol. Exp. Ther. 1999, 288, 714–719. [Google Scholar] [PubMed]
- Schanne, F.A.; Moskal, J.R.; Gupta, P.K. Effect of lead on intracellular free calcium ion concentration in a presynaptic neuronal model: 19F-NMR study of NG108-15 cells. Brain Res. 1989, 503, 308–311. [Google Scholar] [CrossRef]
- Dave, V.; Vitarella, D.; Aschner, J.L.; Fletcher, P.; Kimelberg, H.K.; Aschner, M. Lead increases inositol 1,4,5-trisphospate levels but does not interfere with calcium transient in primary rat astrocytes. Brain Res. 1993, 618, 9–18. [Google Scholar] [CrossRef]
- Gilman, A.G. G-protein: Transducer of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, P.; Di Gioacchino, M.; Sabbioni, E.; Benvenuti, F.; Conti, P.; Reale, M.; Bavazzano, P.; Giuliano, G. Expression of lymphocyte subpopulations, cytokine serum levels, and blood and urinary trace elements in asymptomatic atopic men exposed to an urban environment. Int. Arch. Occup. Environ. Health 1999, 72, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, P.; Di Gioacchino, M.; Spanò, A.; Di Giacomo, F.; Ballone, E.; D’Isidoro, G.; Cavallucci, E.; Giuliano, G. Trace elements in biological samples and immunologic parameters in environmentally exposed populations (preliminary study). Giornale Italiano di Medicina del Lavoro ed Ergonomia 1997, 19, 53–55. [Google Scholar] [PubMed]
- Boscolo, P.; Di Gioacchino, M.; Bavazzano, P.; White, M.; Sabbioni, E. Effects of chromium on lymphocyte subsets and immunoglobulins from normal population and exposed workers. Life Sci. 1997, 60, 1319–1325. [Google Scholar] [CrossRef]
- Victery, W.; Miller, C.R.; Zhu, S.Y.; Goyer, R.A. Effect of different levels and periods of lead exposure on tissue levels and excretion of lead, zinc, and calcium in the rat. Fundam. Appl. Toxicol. 1987, 8, 506–516. [Google Scholar] [CrossRef]
- McCabe, M.J., Jr.; Lawrence, D.A. The heavy metal lead exhibits b cell-stimulatory factor by enhancing B cell Ia expression and differentiation. J. Immunol. 1990, 145, 671–677. [Google Scholar] [PubMed]
- McCabe, M.J., Jr.; Lawrence, D.A. Lead, a major environmental pollutant, is immunomodulatory by its differential effects on CD4+ T cell subsets. Toxicol. Appl. Pharmacol. 1991, 111, 13–23. [Google Scholar] [CrossRef]
- Basaran, N.; Undeger, U. Effects of lead on immune parameters in occupationally exposed workers. Am. J. Ind. Med. 2000, 38, 349–354. [Google Scholar] [CrossRef]
- Mishra, K.P.; Singh, V.K.; Rani, R.; Yadav, V.S.; Chandran, V.; Srivastava, S.P.; Seth, P.K. Effect of lead exposure on the immune response of some occupationally exposed individuals. Toxicology 2003, 188, 251–259. [Google Scholar] [CrossRef]
- Yücesoy, B.; Turhan, A.; Ure, M.; Imir, T.; Karakaya, A. Simultaneous effects of lead and cadmium on NK cell activity and some phenotypic parameters. Immunopharmacol. Immunotoxicol. 1997, 19, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Krocova, Z.; Macela, A.; Kroca, M.; Hernychova, L. The immunomodulatory effect(s) of lead and cadmium on the cells of immune system in vitro. Toxicol. In Vitro 2000, 14, 33–40. [Google Scholar] [CrossRef]
- Flohé, S.B.; Brüggemann, J.; Herder, C.; Goebel, C.; Kolb, H. Enhanced proinflammatory response to endotoxin after priming of macrophages with lead ions. J. Leukoc. Biol. 2002, 71, 417–424. [Google Scholar] [PubMed]
- Lawrence, D.A. In vivo and in vitro effects of lead on humoral and cell-mediated immunity. Infect. Immun. 1981, 31, 136–143. [Google Scholar] [PubMed]
- Horiguchi, S.; Endo, G.; Kiyota, I.; Teramoto, K.; Shinagawa, K.; Wakitani, F.; Tanaka, H.; Konishi, Y.; Kiyota, A.; Ota, A. Frequency of cold infections in workers at a lead refinery. Osaka City Med. J. 1992, 38, 79–81. [Google Scholar] [PubMed]
- Dörpinghaus, M.; Brieger, A.; Panichkina, O.; Rink, L.; Haase, H. Lead ions abrogate lipopolysaccharide-induced nitric monoxide toxicity by reducing the expression of STAT1 and iNOS. J. Trace Elem. Med. Biol. 2016, 37, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Comalada, M.; Valledor, A.F.; Lloberas, J.; López-Soriano, F.; Argilés, J.M.; Bogdan, C.; Celada, A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-α. Blood 2000, 95, 3823–3831. [Google Scholar] [PubMed]
- De la Fuente, H.; Portales-Pérez, D.; Baranda, L.; Díaz-Barriga, F.; Saavedra-Alanís, V.; Layseca, E.; González-Amaro, R. Effect of arsenic, cadmium and lead on the induction of apoptosis of normal human mononuclear cells. Clin. Exp. Immunol. 2002, 129, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Shabani, A.; Rabbani, A. Lead nitrate induced apoptosis in alveolar macrophages from rat lung. Toxicology 2000, 149, 109–114. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Lawrence, D.A. Lead modulation of macrophages causes multiorgan detrimental health effects. J. Biochem. Mol. Toxicol. 2014, 28, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Mondal, T.K.; Lawrence, D.A. Lead effects on development and function of bone marrow-derived dendritic cells promote Th2 immune responses. Toxicol. Appl. Pharmacol. 2007, 222, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Lawrence, D.A. Dendritic cells in immunotoxicity testing. Methods Mol. Biol. 2010, 598, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Aiba, S.; Terunuma, A.; Manome, H.; Tagami, H. Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur. J. Immunol. 1997, 27, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Anetor, J.I.; Adeniyi, F.A. Decreased immune status in Nigerian workers occupationally exposed to lead. Afr. J. Med. Med. Sci. 1998, 27, 169–172. [Google Scholar] [PubMed]
- Sun, L.; Hu, J.; Zhao, Z.; Li, L.; Cheng, H. Influence of exposure to environmental lead on serum immunoglobulin in preschool children. Environ. Res. 2003, 92, 124–128. [Google Scholar] [CrossRef]
- Sarasua, S.M.; Vogt, R.F.; Henderson, L.O.; Jones, P.A.; Lybarger, J.A. Serum immunoglobulins and lymphocyte subset distributions in children and adults living in communities assessed for lead and cadmium exposure. J. Toxicol. Environ. Health Part A 2000, 60, 1–15. [Google Scholar] [PubMed]
- Farkhondeh, T.; Boskabady, M.H.; Kohi, M.K.; Sadeghi-Hashjin, G.; Moin, M. Lead exposure affects inflammatory mediators, total and differential white blood cells in sensitized guinea pigs during and after sensitization. Drug Chem. Toxicol. 2014, 37, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.; Saito, H.; Peebles, R.S., Jr. Advances in mechanisms of allergic disease in 2016. J. Allergy Clin. Immunol. 2017, 140, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.; Lee, W.T.; Lawrence, D.A. In vivo the environmental pollutants lead and mercury induce oligoclonal T cell responses skewed toward type-2 reactivities. Cell. Immunol. 1997, 179, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Bener, A.; Almehdi, A.M.; Alwashc, R. A pilot survey of blood lead levels in various types of workers in the United Arab Emirates. Environ. Int. 2001, 27, 311–314. [Google Scholar] [CrossRef]
- Khazdair, M.R.; Boskabady, M.H.; Afshari, R.; Dadpour, B.; Behforouz, A.; Javidi, M.; Abbasnezhad, A.; Moradi, V.; Tabatabaie, S.S. Respiratory symptoms and pulmonary function tests in lead exposed workers. Iran. Red. Crescent Med. J. 2012, 14, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Boskabady, M.H.; Karimi, G.R.; Samarghandian, S.; Farkhondeh, T. Tracheal responsiveness to methacholine and ovalbumin; and lung inflammation in guinea pigs exposed to inhaled lead after sensitization. Ecotoxicol. Environ. Saf. 2012, 86, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Boskabady, M.H.; Koohi, M.K.; Sadeghi-Hashjin, G.; Moin, M. The effect of lead exposure on selected blood inflammatory biomarkers in guinea pigs. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.D.; Skepper, J.N.; Davenport, A.P. Davenport Detection of endothelin receptors in human coronary artery vascular smooth muscle cells but not endothelial cells by using electron microscope autoradiography. J. Cardiovasc. Pharmacol. 1997, 29, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Haneda, M.; Sawano, H.; Isshiki, K.; Maeda, S.; Koya, D.; Inoki, K.; Yasuda, H.; Kashiwagi, A.; Kikkawa, R. Endothelin-1 induces cyclooxygenase-2 expression via nuclear factor of activated T-cell transcription factor in glomerular mesangial cells. J. Am. Soc. Nephrol. 2001, 12, 1359–1368. [Google Scholar] [PubMed]
- Molero, L.; Farré, J.; García-Mendez, A.; Jiménez Mateos-Caceres, P.; Carrasco, M.C.; Millás, I.; Navarro, F.; Córdoba, M.; Casado, S.; López-Farré, A. Endothelin-1 induced proinflammatory markers in the myocardium and leukocytes of guinea-pigs: Role of glycoprotein IIB/IIIA receptors. Cardiovasc. Res. 2003, 57, 109–118. [Google Scholar] [CrossRef]
- López Farré, A.; Riesco, A.; Espinosa, G.; Digiuni, E.; Cernadas, M.R.; Alvarez, V.; Montón, M.; Rivas, F.; Gallego, M.J.; Egido, J. Effect of endothelin-1 on neutrophil adhesion to endothelial cells and perfused heart. Circulation 1993, 88, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- McCarron, R.M.; Wang, L.; Stanimirovic, D.B.; Spatz, M. Endothelin induction of adhesion molecule expression on human brain microvascular endothelial cells. Neurosci. Lett. 1993, 156, 31–34. [Google Scholar] [CrossRef]
- McMillen, M.A.; Huribal, M.; Cunningham, M.E.; Kumar, R.; Sumpio, B.E. Endothelin-1 increases intracellular calcium in human monocytes and causes production of interleukin-6. Crit. Care Med. 1995, 23, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Khalil-Manesh, F.; Gonick, H.C.; Weiler, E.W.; Prins, B.; Weber, M.A.; Purdy, M.E. Lead-induced hypertension: Possible role of endothelial factors. Am. J. Hypertens. 1993, 6, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Gonick, H.C.; Ding, Y.; Bondy, S.C.; Ni, Z.; Vaziri, N.D. Lead-induced hypertension: Interplay of nitric oxide and reactive oxygen species. Hypertension 1997, 30, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Khalil-Manesh, F.; Gonick, H.C.; Weiler, E.W.J.; Prins, B.; Weber, M.A.; Purdy, R.; Ren, Q. Effect of chelation treatment with dimercaptosuccinic acid (DMSA) on lead-related blood pressure changes. Environ. Res. 1994, 65, 86–99. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metryka, E.; Chibowska, K.; Gutowska, I.; Falkowska, A.; Kupnicka, P.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation. Int. J. Mol. Sci. 2018, 19, 1813. https://doi.org/10.3390/ijms19061813
Metryka E, Chibowska K, Gutowska I, Falkowska A, Kupnicka P, Barczak K, Chlubek D, Baranowska-Bosiacka I. Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation. International Journal of Molecular Sciences. 2018; 19(6):1813. https://doi.org/10.3390/ijms19061813
Chicago/Turabian StyleMetryka, Emilia, Karina Chibowska, Izabela Gutowska, Anna Falkowska, Patrycja Kupnicka, Katarzyna Barczak, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2018. "Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation" International Journal of Molecular Sciences 19, no. 6: 1813. https://doi.org/10.3390/ijms19061813
APA StyleMetryka, E., Chibowska, K., Gutowska, I., Falkowska, A., Kupnicka, P., Barczak, K., Chlubek, D., & Baranowska-Bosiacka, I. (2018). Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation. International Journal of Molecular Sciences, 19(6), 1813. https://doi.org/10.3390/ijms19061813