Mathematical Models in the Description of Pregnane X Receptor (PXR)-Regulated Cytochrome P450 Enzyme Induction

 , , and
, , and
Abstract
1. Introduction
2. Pregnane X Receptor Characterization
2.1. PXR Localization
2.2. PXR Transcriptional Machinery
2.3. PXR Target Genes Regulation
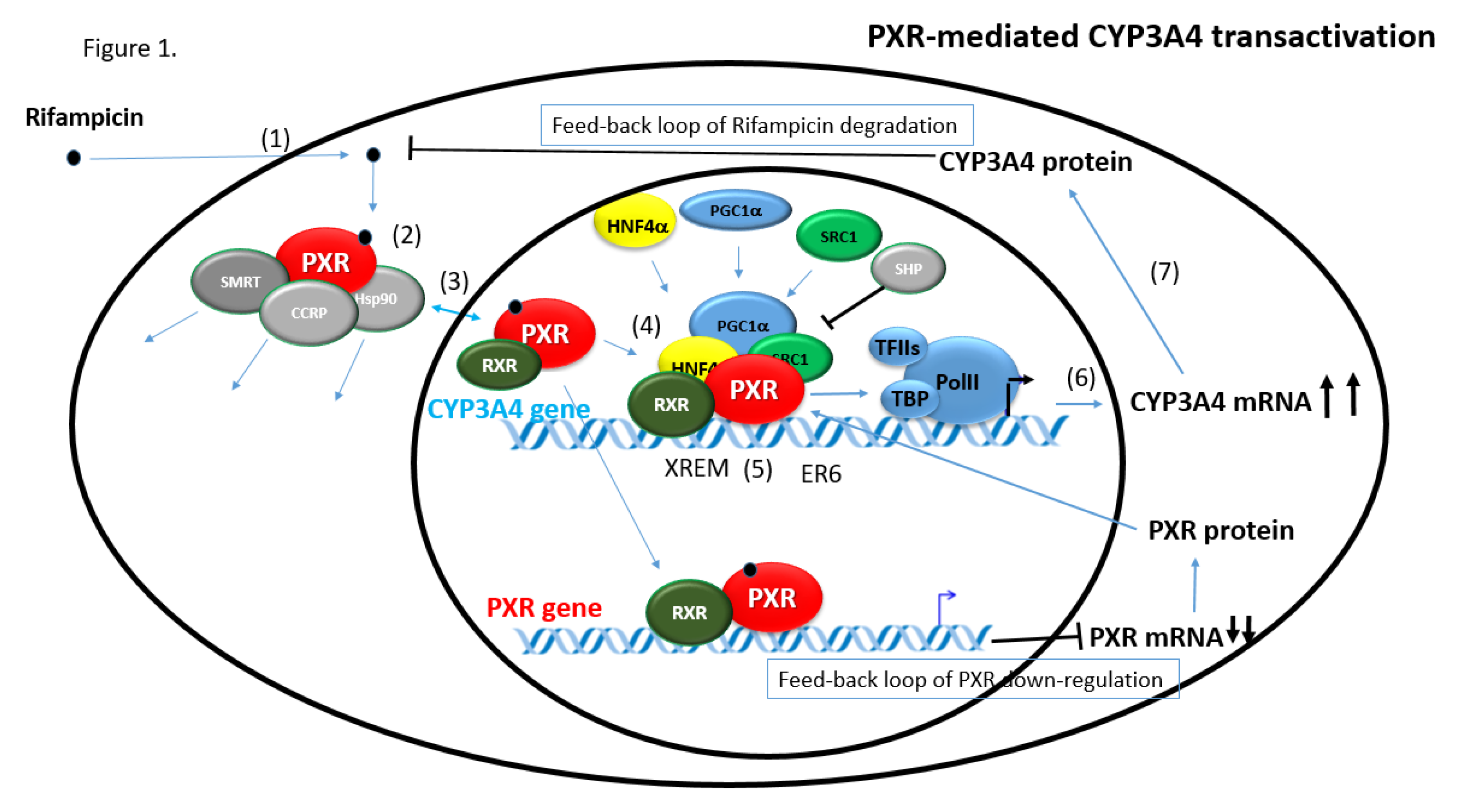
2.3.1. CYP3A4 Gene Regulation via PXR
2.3.2. PXR-Mediated Regulation of Other Target Genes
3. Ligands of the Pregnane X Receptor
4. Brief Overview of Physiological Functions of PXR
5. Mathematical Models of PXR Activation and PXR-Induced Gene Expression
5.1. Compartmental Models
5.2. Models of PXR-Mediated Regulation of CYP3A Enzymes
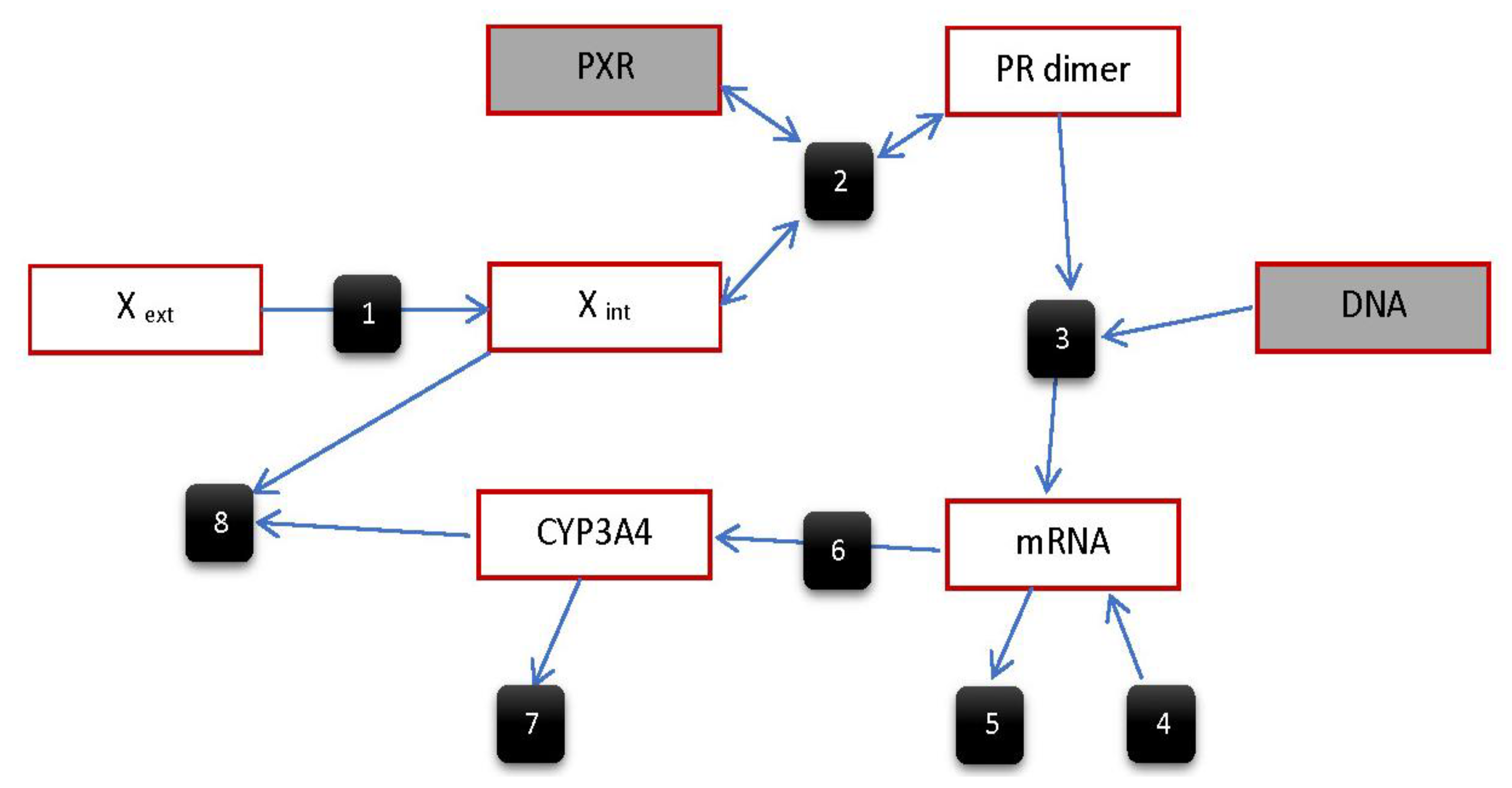
5.2.1. Model by Luke et al.
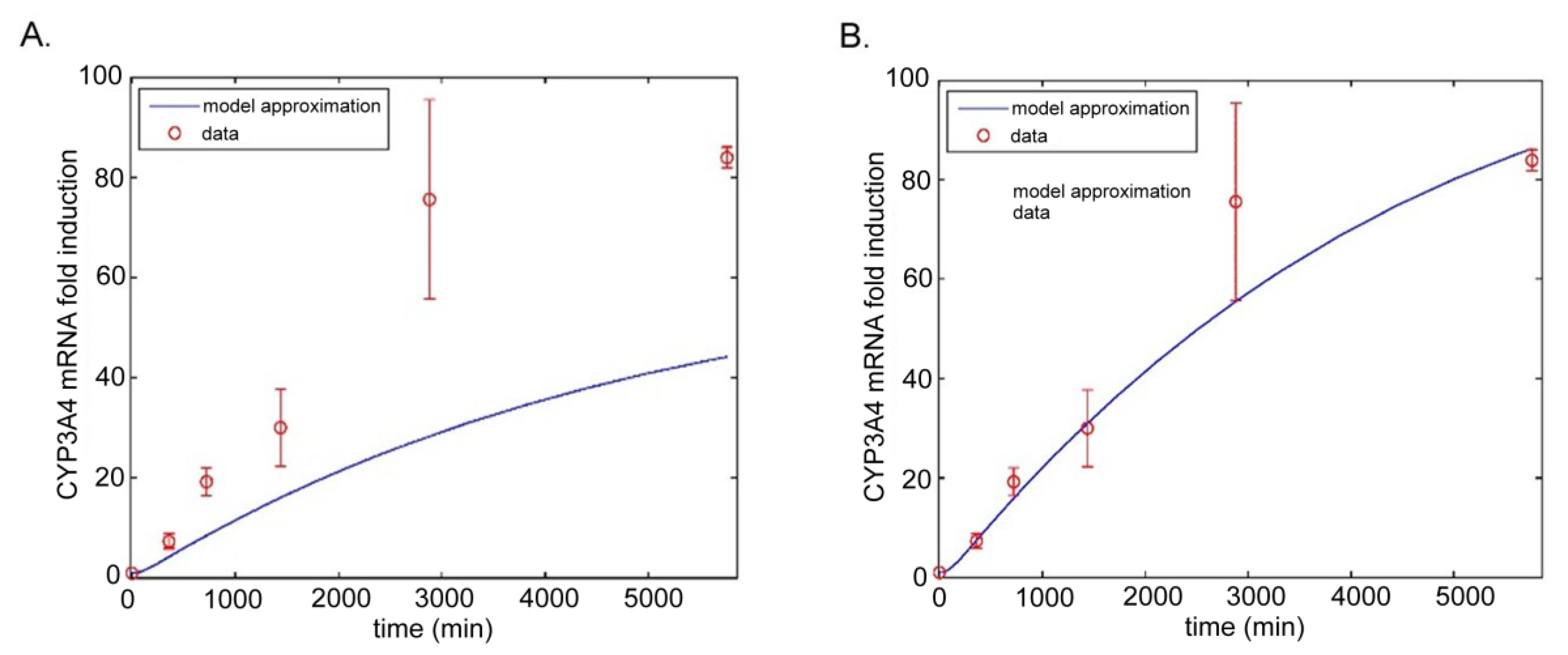
5.2.2. Model by Yamashita et al.
5.2.3. Model by Li et al.
5.2.4. Model by Bailey et al.
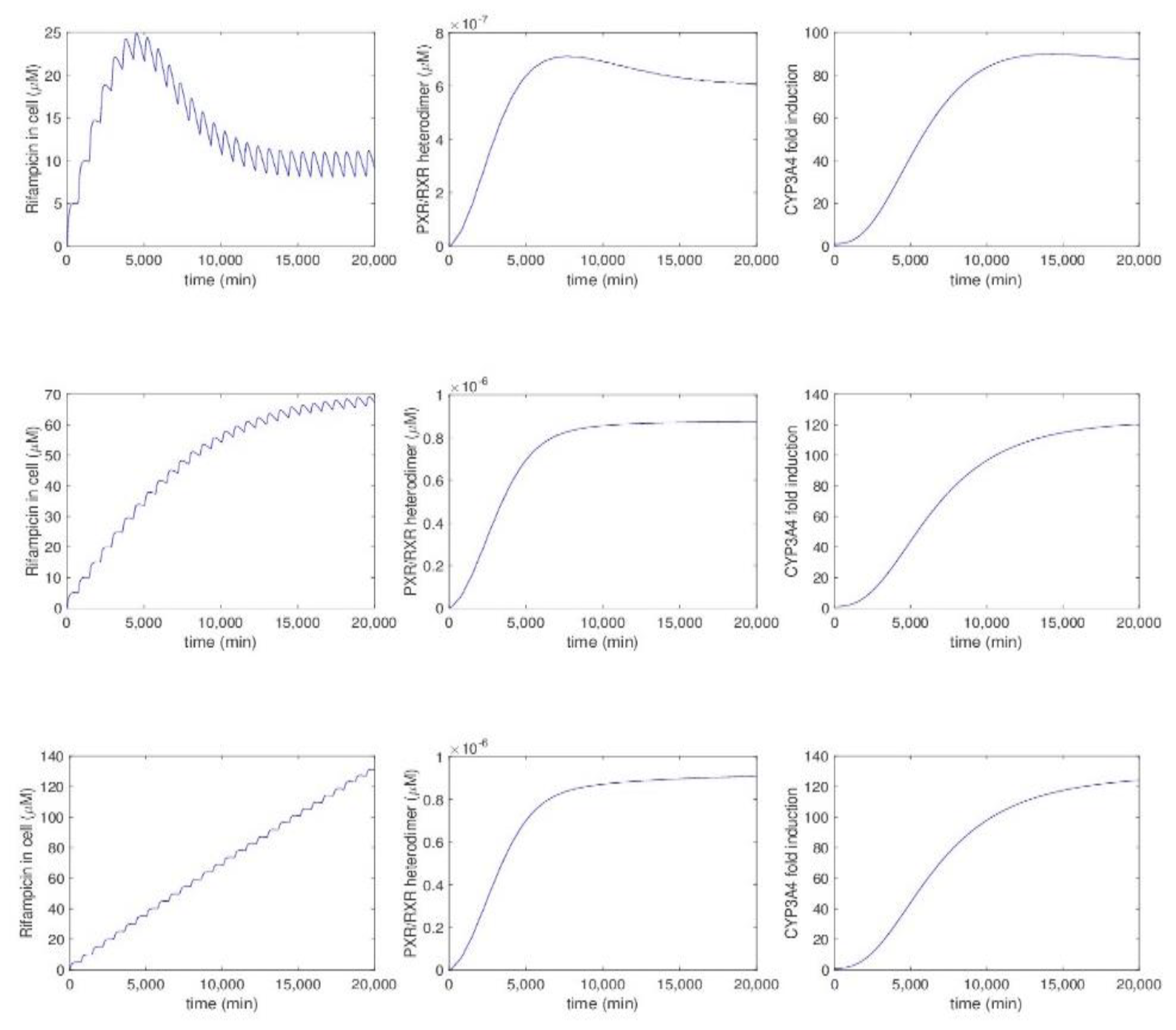
5.2.5. Model by Kolodkin et al.
6. Discussion
6.1. Feedback Loops
6.2. Extended Computational Methodologies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AF-1, AF-2 | Activation function 1, 2 |
| CAR | Constitutive androstane receptor |
| CCRP | Cytoplasmic CAR retention protein |
| CYP | Cytochrome P450 |
| CYP3A4 | Cytochrome P450 3A4 |
| DBD | DNA-binding domain |
| DDI | Drug-drug interaction |
| GR | Glucocorticoid receptor |
| Hsp90 | Heat shock protein 90 |
| LBD | Ligand binding domain |
| NR | Nuclear receptor |
| ODE | Ordinary differential equation |
| PDE | Partial differential equation |
| PK/PD | Pharmacokinetic/pharmacodynamic |
| PAK | Protein kinase A |
| PXR | Pregnane X receptor |
| RXRα | Retinoid X receptor-α |
| SRC | Steroid receptor coactivator |
| TAT | Tyrosine aminotransferase |
| VDR | Vitamin D receptor |
| XREM | Xenobiotic responsive enhancer module |
References
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Baghdasaryan, A.; Trauner, M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 2013, 17, 161–189. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Youn, M.Y.; Inoue, K.; Takada, I.; Kouzmenko, A.; Kato, S. Nuclear receptors in bone physiology and diseases. Physiol. Rev. 2013, 93, 481–523. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Crotti, A.; Glass, C.K. Nuclear receptors, inflammation, and neurodegenerative diseases. Adv. Immunol. 2010, 106, 21–59. [Google Scholar] [PubMed]
- Lazar, M.A. Nuclear hormone receptors: From molecules to diseases. J. Investig. Med. 1999, 47, 364–368. [Google Scholar] [PubMed]
- Di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol. Asp. Med. 2009, 30, 297–343. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.D.; Kato, S.; Xie, W.; Mangelsdorf, D.J.; Schmidt, D.R.; Xiao, R.; Kliewer, S.A. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: Constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol. Rev. 2006, 58, 742–759. [Google Scholar] [CrossRef] [PubMed]
- Smutny, T.; Mani, S.; Pavek, P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr. Drug Metab. 2013, 14, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, B.; Wang, H. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta 2016, 1859, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; di Masi, A.; Trezza, V.; Pallottini, V.; Polticelli, F.; Ascenzi, P. Xenosensors CAR and PXR at work: Impact on statin metabolism. Curr. Drug Metab. 2011, 12, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol. 2016, 7, 456. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B.; Kang, H.; Bolado, J., Jr.; Chen, H.; Craig, A.G.; Moreno, T.A.; Umesono, K.; Perlmann, T.; De Robertis, E.M.; Evans, R.M. BXR, an embryonic orphan nuclear receptor activated by a novel class of endogenous benzoate metabolites. Genes Dev. 1998, 12, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B.; Sabbagh, W., Jr.; Juguilon, H.; Bolado, J., Jr.; van Meter, C.M.; Ong, E.S.; Evans, R.M. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998, 12, 3195–3205. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Dvorak, Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr. Drug Metab. 2008, 9, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A. The nuclear pregnane X receptor regulates xenobiotic detoxification. J. Nutr. 2003, 133 (Suppl. 7), 2444S–2447S. [Google Scholar] [CrossRef] [PubMed]
- Sinz, M.; Kim, S.; Zhu, Z.; Chen, T.; Anthony, M.; Dickinson, K.; Rodrigues, A.D. Evaluation of 170 xenobiotics as transactivators of human pregnane X receptor (hPXR) and correlation to known CYP3A4 drug interactions. Curr. Drug Metab. 2006, 7, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, W.D.; Hassan, H.E.; Wang, H. Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm. Sin. B 2016, 6, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.; Zuniga, B.; Song, C.S.; Jiang, S.; Cropper, J.; Park, S.; Chatterjee, B. Nuclear Receptors in Drug Metabolism, Drug Response and Drug Interactions. Nucl. Recept. Res. 2015, 2, 101178. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Tang, C.; Sant, V.; Li, S.; Poloyac, S.M.; Xie, W. A Molecular Aspect in the Regulation of Drug Metabolism: Does PXR-Induced Enzyme Expression Always Lead to Functional Changes in Drug Metabolism? Curr. Pharmacol. Rep. 2016, 2, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hustert, E.; Zibat, A.; Presecan-Siedel, E.; Eiselt, R.; Mueller, R.; Fuss, C.; Brehm, I.; Brinkmann, U.; Eichelbaum, M.; Wojnowski, L.; et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 1454–1459. [Google Scholar] [PubMed]
- Zhang, J.; Kuehl, P.; Green, E.D.; Touchman, J.W.; Watkins, P.B.; Daly, A.; Hall, S.D.; Maurel, P.; Relling, M.; Brimer, C.; et al. The human pregnane X receptor: Genomic structure and identification and functional characterization of natural allelic variants. Pharmacogenetics 2001, 11, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.T.; Chen, T. PXR variants: The impact on drug metabolism and therapeutic responses. Acta Pharm. Sin. B 2016, 6, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Barwick, J.L.; Quattrochi, L.C.; Mills, A.S.; Potenza, C.; Tukey, R.H.; Guzelian, P.S. Trans-species gene transfer for analysis of glucocorticoid-inducible transcriptional activation of transiently expressed human CYP3A4 and rabbit CYP3A6 in primary cultures of adult rat and rabbit hepatocytes. Mol. Pharmacol. 1996, 50, 10–16. [Google Scholar] [PubMed]
- Yan, J.; Xie, W. A brief history of the discovery of PXR and CAR as xenobiotic receptors. Acta Pharm. Sin. B 2016, 6, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.; Reschly, E.J.; Krasowski, M.D. Functional evolution of the pregnane X receptor. Expert Opin. Drug Metab. Toxicol. 2006, 2, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Scheer, N.; Ross, J.; Kapelyukh, Y.; Rode, A.; Wolf, C.R. In vivo responses of the human and murine pregnane X receptor to dexamethasone in mice. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.B.; Parks, D.J.; Jones, S.A.; Bledsoe, R.K.; Consler, T.G.; Stimmel, J.B.; Goodwin, B.; Liddle, C.; Blanchard, S.G.; Willson, T.M.; et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Barwick, J.L.; Downes, M.; Blumberg, B.; Simon, C.M.; Nelson, M.C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Guzelian, P.S.; Evans, R.M. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 2000, 406, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Kawana, K.; Ikuta, T.; Kobayashi, Y.; Gotoh, O.; Takeda, K.; Kawajiri, K. Molecular mechanism of nuclear translocation of an orphan nuclear receptor, SXR. Mol. Pharmacol. 2003, 63, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Squires, E.J.; Sueyoshi, T.; Negishi, M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J. Biol. Chem. 2004, 279, 49307–49314. [Google Scholar] [CrossRef] [PubMed]
- Saradhi, M.; Sengupta, A.; Mukhopadhyay, G.; Tyagi, R.K. Pregnane and Xenobiotic Receptor (PXR/SXR) resides predominantly in the nuclear compartment of the interphase cell and associates with the condensed chromosomes during mitosis. Biochim. Biophys. Acta 2005, 1746, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lichti, K.; Staudinger, J.L. The mycoestrogen zearalenone induces CYP3A through activation of the pregnane X receptor. Toxicol. Sci. 2006, 91, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Li, C.W.; Chen, L.Y.; Ghosh, J.C.; Chen, J.D. Regulation and binding of pregnane X receptor by nuclear receptor corepressor silencing mediator of retinoid and thyroid hormone receptors (SMRT). Mol. Pharmacol. 2006, 69, 99–108. [Google Scholar] [PubMed]
- Handschin, C.; Meyer, U.A. Induction of drug metabolism: The role of nuclear receptors. Pharmacol. Rev. 2003, 55, 649–673. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Hariparsad, N.; Chu, X.; Yabut, J.; Labhart, P.; Hartley, D.P.; Dai, X.; Evers, R. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009, 37, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Ourlin, J.C.; Lasserre, F.; Pineau, T.; Fabre, J.M.; Sa-Cunha, A.; Maurel, P.; Vilarem, M.J.; Pascussi, J.M. The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity. Mol. Endocrinol. 2003, 17, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Stejskalova, L.; Krausova, L.; Bitman, M.; Vrzal, R.; Dvorak, Z. Rifampicin Does not Significantly Affect the Expression of Small Heterodimer Partner in Primary Human Hepatocytes. Front. Pharmacol. 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Staudinger, J.L. Induction of drug metabolism by forskolin: The role of the pregnane X receptor and the protein kinase a signal transduction pathway. J. Pharmacol. Exp. Ther. 2005, 312, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Staudinger, J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem. Pharmacol. 2005, 69, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Portbury, A.L.; Ronnebaum, S.M.; Zungu, M.; Patterson, C.; Willis, M.S. Back to your heart: Ubiquitin proteasome system-regulated signal transduction. J. Mol. Cell. Cardiol. 2012, 52, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Xu, C.; Biswas, A.; Mani, S. Post-translational modification of pregnane X receptor. Pharmacol. Res. 2011, 64, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Pasquel, D.; Tyagi, R.K.; Mani, S. Acetylation of pregnane X receptor protein determines selective function independent of ligand activation. Biochem. Biophys. Res. Commun. 2011, 406, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tian, L.; Popov, V.M.; Pestell, R.G. Acetylation and nuclear receptor action. J. Steroid Biochem. Mol. Biol. 2011, 123, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Rifampicin induction of CYP3A4 requires pregnane X receptor cross talk with hepatocyte nuclear factor 4alpha and coactivators, and suppression of small heterodimer partner gene expression. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Kandel, B.A.; Thomas, M.; Winter, S.; Damm, G.; Seehofer, D.; Burk, O.; Schwab, M.; Zanger, U.M. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPARalpha in primary human hepatocytes. Biochim. Biophys. Acta 2016, 1859, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Pospechova, K.; Svecova, L.; Syrova, Z.; Stejskalova, L.; Blazkova, J.; Dvorak, Z.; Blahos, J. Intestinal cell-specific vitamin D receptor (VDR)-mediated transcriptional regulation of CYP3A4 gene. Biochem. Pharmacol. 2010, 79, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Moore, L.B.; Stoltz, C.M.; McKee, D.D.; Kliewer, S.A. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol. Pharmacol. 2001, 60, 427–431. [Google Scholar] [PubMed]
- Gerbal-Chaloin, S.; Pascussi, J.M.; Pichard-Garcia, L.; Daujat, M.; Waechter, F.; Fabre, J.M.; Carrere, N.; Maurel, P. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 242–251. [Google Scholar] [PubMed]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef] [PubMed]
- Light, R.W.; Lee, Y.C.G. Textbook of Pleural Diseases, 3rd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2016; 651p. [Google Scholar]
- Staudinger, J.; Liu, Y.; Madan, A.; Habeebu, S.; Klaassen, C.D. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 1467–1472. [Google Scholar] [PubMed]
- Teng, S.; Jekerle, V.; Piquette-Miller, M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, G.; Mnif, W.; Pascussi, J.M.; Pillon, A.; Rabenoelina, F.; Fenet, H.; Gomez, E.; Casellas, C.; Nicolas, J.C.; Cavailles, V.; et al. Identification of new human pregnane X receptor ligands among pesticides using a stable reporter cell system. Toxicol. Sci. 2006, 91, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Redinbo, M.R.; Kliewer, S.A. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.E.; Maglich, J.M.; Moore, L.B.; Wisely, G.B.; Noble, S.M.; Davis-Searles, P.R.; Lambert, M.H.; Kliewer, S.A.; Redinbo, M.R. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry 2003, 42, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.J.; Rahman, S.R.; Levy, C.; Hirschfield, G.M. Low risk of hepatotoxicity from rifampicin when used for cholestatic pruritus: A cross-disease cohort study. Aliment. Pharmacol. Ther. 2018, 47, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Slitt, A.L. Regulation of hepatic transporters by xenobiotic receptors. Curr. Drug Metab. 2005, 6, 309–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, S.; Guo, Y.; Xie, W.; Zhai, Y. Biology of PXR: Role in drug-hormone interactions. EXCLI J. 2014, 13, 728–739. [Google Scholar] [PubMed]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grun, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xie, W.; Krasowski, M.D. PXR: A xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 2008, 9, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Pascussi, J.M.; Robert, A.; Nguyen, M.; Walrant-Debray, O.; Garabedian, M.; Martin, P.; Pineau, T.; Saric, J.; Navarro, F.; Maurel, P.; et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J. Clin. Investig. 2005, 115, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G. Vitamin E, nuclear receptors and xenobiotic metabolism. Arch. Biochem. Biophys. 2004, 423, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Landes, N.; Pfluger, P.; Kluth, D.; Birringer, M.; Ruhl, R.; Bol, G.F.; Glatt, H.; Brigelius-Flohe, R. Vitamin E activates gene expression via the pregnane X receptor. Biochem. Pharmacol. 2003, 65, 269–273. [Google Scholar] [CrossRef]
- Ruhl, R. Induction of PXR-mediated metabolism by beta-carotene. Biochim. Biophys. Acta 2005, 1740, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Tian, Y. Xenobiotic receptor meets NF-kappaB, a collision in the small bowel. Cell Metab. 2006, 4, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grun, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Pondugula, S.R.; Pavek, P.; Mani, S. Pregnane X Receptor and Cancer: Context-Specificity is Key. Nucl. Recept. Res. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Rysa, J.; Hukkanen, J. Regulation of hepatic energy metabolism by the nuclear receptor PXR. Biochim. Biophys. Acta 2016, 1859, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Oladimeji, P.O.; Chen, T. PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharmacol. 2018, 93, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.T.T. Clinical Pharmacokinetics: Concepts and Applications; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Gesheff, M.G.; Franzese, C.J.; Bliden, K.P.; Contino, C.J.; Rafeedheen, R.; Tantry, U.S.; Gurbel, P.A. Review of pharmacokinetic and pharmacodynamic modeling and safety of proton pump inhibitors and aspirin. Expert Rev. Clin. Pharmacol. 2014, 7, 645–653. [Google Scholar] [CrossRef] [PubMed]
- De Jong, H.; Ropers, D. Strategies for dealing with incomplete information in the modeling of molecular interaction networks. Brief. Bioinform. 2006, 7, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; DuBois, D.C.; Almon, R.R.; Pyszczynski, N.A.; Jusko, W.J. Fifth-generation model for corticosteroid pharmacodynamics: Application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J. Pharmacokinet. Pharmacodyn. 2002, 29, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ayyar, V.S.; Almon, R.R.; Jusko, W.J.; DuBois, D.C. Quantitative tissue-specific dynamics of in vivo GILZ mRNA expression and regulation by endogenous and exogenous glucocorticoids. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Ayyar, V.S.; Sukumaran, S.; DuBois, D.C.; Almon, R.R.; Qu, J.; Jusko, W.J. Receptor/gene/protein-mediated signaling connects methylprednisolone exposure to metabolic and immune-related pharmacodynamic actions in liver. J. Pharmacokinet. Pharmacodyn. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Dutreix, C.; Jarugula, V.; Rebello, S.; Won, C.S.; Sun, H. An Exposure-Response Modeling Approach to Examine the Relationship between Potency of CYP3A Inducer and Plasma 4beta-Hydroxycholesterol in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2017, 6, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Leil, T.A.; Kasichayanula, S.; Boulton, D.W.; LaCreta, F. Evaluation of 4beta-Hydroxycholesterol as a Clinical Biomarker of CYP3A4 Drug Interactions Using a Bayesian Mechanism-Based Pharmacometric Model. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e120. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Tagawa, Y.; Kimura, Y.; Kogame, A.; Moriya, Y.; Amano, N. Influence of the pharmacokinetic profile on the plasma glucose lowering effect of the PPARgamma agonist pioglitazone in Wistar fatty rats. Biopharm. Drug Dispos. 2017, 38, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Jin, Y.; Li, Y.G.; Borel, A. Population pharmacokinetic/pharmacodynamic assessment of pharmacological effect of a selective estrogen receptor beta agonist on total testosterone in healthy men. Clin. Pharmacol. Drug Dev. 2015, 4, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, F.; Sasa, Y.; Yoshida, S.; Hisaka, A.; Asai, Y.; Kitano, H.; Hashida, M.; Suzuki, H. Modeling of rifampicin-induced CYP3A4 activation dynamics for the prediction of clinical drug-drug interactions from in vitro data. PLoS ONE 2013, 8, e70330. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin, A.; Sahin, N.; Phillips, A.; Hood, S.R.; Bruggeman, F.J.; Westerhoff, H.V.; Plant, N. Optimization of stress response through the nuclear receptor-mediated cortisol signalling network. Nat. Commun. 2013, 4, 1792. [Google Scholar] [CrossRef] [PubMed]
- Luke, N.S.; DeVito, M.J.; Shah, I.; El-Masri, H.A. Development of a quantitative model of pregnane X receptor (PXR) mediated xenobiotic metabolizing enzyme induction. Bull. Math. Biol. 2010, 72, 1799–1819. [Google Scholar] [CrossRef] [PubMed]
- Bailey, I.; Gibson, G.G.; Plant, K.; Graham, M.; Plant, N. A PXR-mediated negative feedback loop attenuates the expression of CYP3A in response to the PXR agonist pregnenalone-16alpha-carbonitrile. PLoS ONE 2011, 6, e16703. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Panetta, J.C.; Strom, S.; Schuetz, E.G. Genetic predictors of interindividual variability in hepatic CYP3A4 expression. J. Pharmacol. Exp. Ther. 2010, 332, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Slatter, J.G.; Templeton, I.E.; Castle, J.C.; Kulkarni, A.; Rushmore, T.H.; Richards, K.; He, Y.; Dai, X.; Cheng, O.J.; Caguyong, M.; et al. Compendium of gene expression profiles comprising a baseline model of the human liver drug metabolism transcriptome. Xenobiotica 2006, 36, 938–962. [Google Scholar] [CrossRef] [PubMed]
- Kotta-Loizou, I.; Patsouris, E.; Theocharis, S. Pregnane X receptor polymorphisms associated with human diseases. Expert Opin. Ther. Targets 2013, 17, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Devi, S.; Gourinath, S.; Goswami, R.; Tyagi, R.K. A comprehensive analysis and functional characterization of naturally occurring non-synonymous variants of nuclear receptor PXR. Biochim. Biophys. Acta 2016, 1859, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Z.Q.; Deng, C.H.; Ning, M.R.; Li, H.Q.; Bi, S.S.; Zhou, T.Y.; Lu, W. A mechanism-based pharmacokinetic/pharmacodynamic model for CYP3A1/2 induction by dexamethasone in rats. Acta Pharmacol. Sin. 2012, 33, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Harley, E.M.; Loftus, G.R. MATLAB and graphical user interfaces: Tools for experimental management. Behav. Res. Methods Instrum. Comput. 2000, 32, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Matsuoka, Y.; Asai, Y.; Hsin, K.Y.; Kitano, H. Software for systems biology: From tools to integrated platforms. Nat. Rev. Genet. 2011, 12, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Sheiner, L.B.; Grasela, T.H. Experience with NONMEM: Analysis of routine phenytoin clinical pharmacokinetic data. Drug Metab. Rev. 1984, 15, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Pascussi, J.M.; Drocourt, L.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: Synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol. Pharmacol. 2000, 58, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Homologous down regulation of the glucocorticoid receptor: The molecular machinery. Crit. Rev. Eukaryot. Gene Expr. 1993, 3, 63–88. [Google Scholar] [PubMed]
- Rigaud, G.; Roux, J.; Pictet, R.; Grange, T. In vivo footprinting of rat TAT gene: Dynamic interplay between the glucocorticoid receptor and a liver-specific factor. Cell 1991, 67, 977–986. [Google Scholar] [CrossRef]
- Plant, N. The human cytochrome P450 sub-family: Transcriptional regulation, inter-individual variation and interaction networks. Biochim. Biophys. Acta 2007, 1770, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Raybon, J.J.; Pray, D.; Morgan, D.G.; Zoeckler, M.; Zheng, M.; Sinz, M.; Kim, S. Pharmacokinetic-pharmacodynamic modeling of rifampicin-mediated Cyp3a11 induction in steroid and xenobiotic X receptor humanized mice. J. Pharmacol. Exp. Ther. 2011, 337, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.G.; Phillips, A.; Aouabdi, S.; Plant, K.; Plant, N. Transcriptional regulation of the human pregnane-X receptor. Drug Metab. Rev. 2006, 38, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, J.L. Microcomputer-assisted kinetic modeling of mammalian gene expression. FASEB J. 1993, 7, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Claus, J.; Friedmann, E.; Klingmuller, U.; Rannacher, R.; Szekeres, T. Spatial aspects in the SMAD signaling pathway. J. Math. Biol. 2013, 67, 1171–1197. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, E. PDE/ODE modeling and simulation to determine the role of diffusion in long-term and -range cellular signaling. BMC Biophys. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Thurley, K.; Gerecht, D.; Friedmann, E.; Hofer, T. Three-Dimensional Gradients of Cytokine Signaling between T Cells. PLoS Comput. Biol. 2015, 11, e1004206. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands with High Potency | Ligands with Medium Potency | Ligands with Low Potency |
|---|---|---|
| Rifampicin | Carbamazepine | Testosterone |
| Mifepristone | Dexamethasone | Indomethacin |
| SR12813 | Zolpidem | Warfarin |
| Hyperforin | Rosiglitazone | Ethinyl estradiol |
| Nifedipine | Atorvastatin | Phenobarbital |
| Diltiazem | Omeprazole | Rosuvastatin |
| Clotrimazole | Loratadine | Tamoxifen |
| Rifabutine | Haloperidole | Saquinavir |
| Authors | PXR Ligand | Nuclear Receptor | PXR Target Gene Studied | Tested Model | Software Used | Data Sets |
|---|---|---|---|---|---|---|
| [90] | Rifampicin | PXR | CYP3A4 | - Primary human hepatocytes - Two compartment model of with human central compartment and the liver (livers scaled from hepatocytes in culture using a scaling factor) | MATLAB | - CYP3A4 mRNA data in primary human hepatocytes from literature - human plasma concentration of rifampicin from literature |
| [88] | Rifampicin | PXR | CYP3A4 | - Primary human hepatocytes - two compartment PBPK model | CellDesigner, PhysioDesigner, NONMEM | - CYP3A4 mRNA data in primary human hepatocytes from literature - human plasma concentration of rifampicin |
| [96] | Dexamethasone (a rat PXR ligand) | PXR | CYP3A1/2 (a rat CYP3A4 orthologue) | Rats - PK/PD model after i.p. application | NONMEM | Animal experiments, pharmacokinetic study with Sprague-Dawley rats receiving 100 mg/kg of dexamenthasone i.p. |
| [91] | PCN, LCA (rat PXR ligands) | PXR (VDR, FXR) | CYP3A1, CYP24, Fibrinogen B | Rats and human cell line Huh7 | CellDesigner, MVSP | primary rat hepatocytes and Huh7 cells |
| [89] | Cortisol | PXR, GR | CYP3A4, TAT | Humans | CellDesigner | - primary human hepatocytes - data from literature |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duintjer Tebbens, J.; Azar, M.; Friedmann, E.; Lanzendörfer, M.; Pávek, P. Mathematical Models in the Description of Pregnane X Receptor (PXR)-Regulated Cytochrome P450 Enzyme Induction. Int. J. Mol. Sci. 2018, 19, 1785. https://doi.org/10.3390/ijms19061785
Duintjer Tebbens J, Azar M, Friedmann E, Lanzendörfer M, Pávek P. Mathematical Models in the Description of Pregnane X Receptor (PXR)-Regulated Cytochrome P450 Enzyme Induction. International Journal of Molecular Sciences. 2018; 19(6):1785. https://doi.org/10.3390/ijms19061785
Chicago/Turabian StyleDuintjer Tebbens, Jurjen, Malek Azar, Elfriede Friedmann, Martin Lanzendörfer, and Petr Pávek. 2018. "Mathematical Models in the Description of Pregnane X Receptor (PXR)-Regulated Cytochrome P450 Enzyme Induction" International Journal of Molecular Sciences 19, no. 6: 1785. https://doi.org/10.3390/ijms19061785
APA StyleDuintjer Tebbens, J., Azar, M., Friedmann, E., Lanzendörfer, M., & Pávek, P. (2018). Mathematical Models in the Description of Pregnane X Receptor (PXR)-Regulated Cytochrome P450 Enzyme Induction. International Journal of Molecular Sciences, 19(6), 1785. https://doi.org/10.3390/ijms19061785